

- • DEFINITIONS
- • DRUG NOMENCLATURE
- • PHARMACOPOEIA
- • SOURCES OF DRUGS
- • ROUTES OF DRUG ADMINISTRATION
Pharmacology is the science of drugs. Like many other subjects, pharmacology had its humble origin thousands of years ago. It is as old as man himself, since, fighting diseases with remedies appear to be an integral part of human life. Search for drugs useful in diseases is a timeless struggle. The rational approach to this struggle is pharmacology. Pharmakon is a Greek term which means “drug” and logos means a systematic study. Modern pharmacology is an ever-expanding discipline of science. With the advent of new techniques of understanding of human physiology and biochemistry, pharmacology is growing at a rapid rate. Further, attempts to reduce adverse effects of drugs have been successful and this has made modern medicine more popular. Method of administration, new drug delivery systems, proven drug efficacy and quality improvement has resulted in increased rate of success in the control of many chronic illnesses. Clinicians, manufacturers of drugs and drug sellers must be aware of the rationale of drug therapy in order to avoid any drug-induced disaster. Physiotherapy is a well-accepted mode of re-equipping the patient back to a normal productive life. A clear knowledge of commonly used drugs is essential for a physiotherapist in view of achieving expected success in the field.
DEFINITIONS
Drug [Drogue (French): dry herb]
The World Health Organization defines drug as “any substance or product that is used or intended to be used to modify or explore physiological system or pathological state for the benefit of the recipient.”
Drug is a substance used in prevention, diagnosis, treatment and cure of diseases. With obvious exceptions, drugs are xenobiotic (xenos: strange, foreign) to human body and hence 2prone to induce allergic reactions in many individuals. This suggests that interaction of drugs and living organisms constitutes the main basis for beneficial or harmful drug effects.
Pharmacokinetics
Pharmacokinetics is that branch of pharmacology which deals with the study of absorption, distribution, metabolism and excretion of drugs in a biological system.
Pharmacodynamics
Pharmacodynamics deals with the study of the actions and effects of drugs at all levels of interaction in a biological system. This describes the actions and mechanisms of the action of drugs.
Pharmacotherapeutics
This correlates the principles of pharmacodynamics with pathologic physiology or microbiological or biochemical aspects of disease. It is the application of concepts of pharmacology to a clinical condition.
Toxicology
Toxicology is the science of poisons. It is the study of harmful effects of chemicals as well as occurrence, mechanisms, conditions, management, and treatment of adverse effects.
Environmental toxicology deals with incidental or occupational hazards caused by chemicals of the atmosphere, water and food.
DRUG NOMENCLATURE
Drugs used as therapeutic agents are grouped into two broad classes:
- Over-the-counter drugs (OTC): Non-prescription drugs which are judged safe and for use without medical supervision.
- Prescription drugs: To be used under medical supervision. Therefore, dispensed only by the order of licensed practitioners, physicians, dentists and veterinarians.
Drugs may also be referred by either chemical or pharmacological relationship among a group of agents. This contributes to their “Generic name”. For example, benzodiazepines, thiazides, opioids, cardiac glycosides, loop diuretics, etc. As a whole, generic name of drugs focus on the pharmacological similarities among the members of a class.
Further, therapeutic agents at least have 3 names. Every drug has a chemical name, a non-proprietary name and a proprietary or trade name or brand name. The non-proprietary name is frequently referred to as the generic name of the drug. By strict definition, this is inappropriate. However, it should be reserved to designate a family relationship among drugs as said above.
Chemical name is the first name of a drug, which indicates the chemical constitution, and arrangements of atoms or atomic groups, for example, 3-4-dihydroxyphenylethylamine–catecholamine–adrenaline. The non-proprietary name of a drug is the name assigned to it when it is said to have demonstrated therapeutic potential, for example, chlordiazepoxide–3which is a benzodiazepine anti-anxiety drug. The non-proprietary name when finally admitted to official compound becomes an approved official name of the drug.
The trade name or brand name is selected by the pharmaceutical company. For example, trade name for chlordiazepoxide–LIBRIUM, diazepam–CALMPOSE, paracetamol–METACIN, CROCIN. As a rule, the proprietary (brand or trade) name is shorter, more euphonic and easier to recall than the non-proprietary name. Now, major countries have a national council for giving adapted names to drugs. A single drug may have many different trade names. Often this brings confusion, which is generally minimized by furnishing non-proprietary name along with the trade name and this ensures accurate recognition of an agent.
A number of compendia at the national and international levels have been published annually at a regular time interval to meet the need of current, critical sources of drug information. There are also several publications, which furnish recent developments in drug therapy and toxicity.
PHARMACOPOEIA
Pharmacopoeia is an official compendium containing selected lists of approved drugs with uses, dose, method of administration, toxicities, drug interactions, purity, potency and tests for identification, for example, USP: United States Pharmacopoeia, BP: British Pharmacopoeia, IP: Indian Pharmacopoeia.
SOURCES OF DRUGS
Drugs have been obtained from the following sources:
- plant source
- synthetic chemical laboratory
- animal source
- minerals
- microbiological source
Plant Source
From time immemorial, drugs have been extracted from plants. In fact, the word ‘drug’ took its origin from the French word drogue meaning dry herb. Modern medicine now has accepted various herbal medicines in its drug armamentarium. Drugs obtained from plants can be grouped as:
- Alkaloids (alkali-like): Nitrogenous bases which produce marked effects in a living organism. These form water-soluble salts with acid, for example, morphine, atropine, caffeine and quinine.
- Glycosides: Naturally occurring substances, which represent the combination of a non-sugar moiety called ‘aglycone’ with 3 to 4 molecules of sugar. Glycosides on acid hydrolysis liberate reducible sugars, for example, digoxin and digitoxin.
Other plant products include oils, tannins, gums, resins and saponins, which are employed as drugs in various conditions, and are also being used in the drug manufacturing industry.4
Animal Source
Various drugs are obtained from animals, for example, insulin, heparin and protamine. Different hormones have been isolated from various animals for the treatment of many endocrinal disorders.
Minerals
Iron, magnesium, iodine, phosphorus, gold and their salts have been used in modern medicine. Gold salts in rheumatoid arthritis and magnesium salts in hyperacidity and acid peptic disease are a few examples for mineral drug use.
Synthetic Drugs
Organic chemical synthetic laboratory is a major source for drugs today. Chemical structure of a drug is intimately associated with the action. Any modification of drug structure results in qualitative changes in the pharmacodynamic as well as kinetic profiles besides toxicity change. Therefore, it is possible to synthesize various structural analogues of established drugs to mitigate adverse effects and increase potency, purity and safety of drugs. By this process, the advent of new drugs is a continuing process since the quest for newer, safer drugs is a timeless, limitless demand. Of late, recombinant DNA technology has revolutionized the art of drug production in the laboratory. More purified forms of human insulin is being produced by this technique. Drugs produced by recombinant DNA technology offer many advantages in the purity, immunogenicity and potency although remain expensive.
Microbiological Source
A wide variety of antibiotics have been isolated from different species of bacteria, for example, penicillins, cephalosporins, tetracyclines. However, semisynthetic derivatives of antibiotics are being produced in the laboratory after the elucidation of the structure of an antibiotic, for example, aminopenicillins.
ROUTES OF DRUG ADMINISTRATION
Drugs are administered by several routes. Each route offers advantages and has limitations. The selection of route of drug administration often demands a thorough consideration of physical and chemical properties of drugs and patient characteristics. At times, the chosen drug dosage form does indicate the route of administration. Hence, the knowledge about the selected route is necessary for both patient and practitioner. This often helps to reduce the cost of drug treatment, for example, oral preparations are comparatively cheaper than parenteral preparations.
The major routes of administration are as follows:
Local
- Application
- Inhalation
- Inunction
- Instillation
- Insufflation
- Iontophoresis
Enteral
- Sublingual
- Oral
- Rectal
Parenteral
Intradermal | Intraventricular | Intraureteral |
Subcutaneous | Intracardiac | Intracisternal |
Intramuscular | Intracoronary | Intravitreal |
Intravenous | Intravaginal | Intralesional |
Intra-arterial | Intra-amniotic | Intraneural |
Intrathecal | Intra-bone marrow | Epidural |
Intraperitoneal | Intracavernous | |
Intrapleural | Intraarticular |
Transdermal
Drugs are applied to skin for systemic action. Transdermal use of drugs minimizes adverse drug reactions and takes away repeated oral consumption of drugs. More importantly, a steady state concentration of drug in plasma is maintained. Scopolamine, clonidine and nitroglycerin are available as transdermal preparations.
Local
Drugs are used locally to produce actions at the site of administration. There are many ways and methods for local use of drugs.
Application
Drugs are applied to mucous membrane or skin to produce local action at the applied site, e.g. surface anesthetics. Anti-inflammatory, antimicrobial and antiallergic drugs are used in the form of ointment, cream, jelly and lotions.
Instillation
Drug solutions are instilled to conjunctival sac or nasal cavity or any other body cavities, for example, eyedrops or nasal drops.
Insufflation
Insufflation is a method of dressing body cavities with powder form of drugs, for example, nebasulpha insufflation.
Inhalation
Various drugs are administered by inhalation to elicit the action at the desired site promptly, for example, salbutamol inhalation for bronchial asthma and insulin nasal spray to reduce postprandial elevation of blood sugar.6
Inunction
This is a method of application of drugs on unbroken skin with thorough rubbing or friction. Counter-irritant preparations are applied by inunction to provide relief from pain.
Iontophoresis
A wave of Galvanic current is applied along with the application of drugs on skin to promote dermal absorption. This is a special technique of dermal application of drugs.
Enteral Routes
Sublingual (Buccal)
This route is often employed to produce drug action rapidly within a few seconds to minutes in an ambient patient. Drug is placed beneath the tongue to have prompt action and when the drug action is no longer required, the tablet can be spit out. The drug dosage forms meant for sublingual administration are either known as buccal tablets or buccal sprays. Very few drugs are given by this route—nitroglycerin in angina pectoris, buprenorphine for pain relief, nifedipine in hypertensive crisis.
Sublingual administration of drugs produce rapid action and the action of the drug can be terminated at will by spitting out the tablet. Importantly, drugs administered by sublingual route bypass the liver and enter the circulation directly. Therefore, drugs, which undergo extensive first pass metabolism on oral administration, can be conveniently administered by sublingual route.
The major limitations of this route are irritant and unpleasant drugs cannot be given. Significantly, length of time needed for drug administration is limited. Besides, buccal route cannot be employed when the patient is unco-operative, and while doing exercise.
Oral Route (PO: Per oral)
Most commonly drugs are administered by mouth since it offers many advantages. Oral route does not require special purification procedures for drug dosage forms. Method of administration is safe, convenient and economical. Self-medication is possible with oral route. However, irritant substances are not suitable for oral route. This is not the route for emergency situations. Administration of drugs is not possible in an unconscious patient. If the patient is unco-operative, it is better not to employ oral route. Drugs which are destroyed by proteolytic and other enzymes of gastrointestinal tract cannot be given by mouth, for example, insulin and adrenaline. Presence of food, motility of the alimentary tract to a larger extent, alters the route of oral absorption of drugs. For example, an antitubercular drug rifampicin is better absorbed when the stomach is empty, whereas griseofulvin, an antifungal drug is well absorbed after food. Further, drugs which undergo complete metabolism before reaching circulation, cannot be given by oral route, for example, lignocaine. Oral administration is most suitable for long-term drug treatments.
Rectal Administration (PR: Per rectum)
Drugs can be administered by rectal route even when the patient is unconscious. Vomiting does not preclude rectal administration of drugs. In fact, gastric mucosal irritant drugs like 7indomethacin are given by rectal route. There are two different types of drug dosage forms meant for rectal administration:
- Suppository
- Enema
A suppository is a cone-shaped solid containing medicaments meant for rectal administration. Suppository, which is in solid state at room temperature, melts at body temperature. These preparations contain greasy or non-greasy bases. Examples of drugs which are available as suppository are bisacodyl (a purgative), diazepam (a sedative-hypnotic), indomethacin (antiinflammatory analgesic), glycerine + senna (a purgative combination).
The liquid meant for rectal administration is known as “enema”. Mainly two types of enema are in use:
- Retention enema, for example, prednisolone enema used in ulcerative colitis
- Evacuant enema, for example, soap water enema commonly given before surgery to evacuate the bowel.
Drugs, which are absorbed from the lowest part of rectum, do escape hepatic metabolism and directly enter circulation. However, drug absorption across rectal mucosa is irregular and incomplete.
Parenteral Routes
Routes other than enteral routes are known as parenteral routes. Drugs administered by parenteral routes require aseptic measures and are often given as injectable solutions. Most commonly, subcutaneous, intramuscular and intravenous parenteral routes are employed. The solid medicament meant for subcutaneous administration is known as pellet. Pellets are implanted subcutaneously to produce action for a few days to months. Steroidal drugs are given as pellets. The quantity of solution given by intramuscular route should not exceed 10 ml at a time.
Intravenous Route of Administration
Intravenous route is the route of emergency. Drugs are introduced directly into the vascular compartment. The mode of intravenous administration of drugs differs according to the need in a given clinical condition. Drugs can be given intravenously either as bolus injection or jet injection or infusion. Bolus injection means that the administration of dose of a drug all at once. Adenosine is given as bolus injection in the treatment of paroxysmal supraventricular tachycardia. Administration of drugs by intravenous route at a required pace is called infusion. Generally, to avoid complications, drugs are infused slowly into the vascular compartment.
Intravenous drug administration offers many advantages—rapid onset of action, any amount of the drug can be given and the rate of administration of a drug can be regulated as required. Since drug is introduced into the circulation, factors that modify absorption of drugs are being circumvented. However, intravenous route is not free from limitations. It requires absolute aseptic measures and skill. Intravenous injectable preparations are expensive. Once the drug is introduced, no retrieval can be done. On intravenous administration of drugs, the individual is more prone for drug toxicity in an unpredictable way. Drugs which are exclusively given by intravenous route are sodium nitroprusside in hypertensive crisis and plasma expanders in hypovolemic shock.8
PHARMACOKINETICS
INTRODUCTION
Drugs must move from where they are administered to the site of action. Drugs to reach their locus of action must pass through various cells and tissues that act as barriers to the migration of materials including drugs are called “biological barriers” (i.e. semipermeable cell membranes). Biological barriers are of different types (namely, cell membrane, blood-brain barrier, placental barrier, dermal barrier). Although they differ in their structure, basically, behave as semipermeable barriers. Some biological barriers allow certain chemicals to pass freely, others allow to pass with difficulty and still others restrict the biotransport almost entirely. The solubility of biotransport materials across the anatomical barrier is the consequence of the physico-chemical properties and structural configuration of the barrier as well of the drug molecule.
Pharmacokinetics is that branch of pharmacology, which deals with drug absorption, distribution, metabolism and excretion. The knowledge of drug kinetics in the ailing human body is essential for clinicians to individualize and optimize the drug dosage regimens. Further, how disease affects pharmacokinetics of the given drug in a patient is necessary to avoid complications of drug therapy.9
DRUG ABSORPTION
The transport of drugs from the site of administration to systemic circulation is known as drug absorption. The rate of drug absorption determines the intensity and duration of action. Therefore an administered drug must cross various cellular and subcellular structures to reach its site of action. Drugs applied on skin or any surface must cross surface barriers, whereas orally administered drugs have to pass through alimentary tract to reach circulation. Despite the anatomical differences of different absorbing surfaces, transport of drugs across these barriers appears to be remarkably similar.
The permeation of substances across simple biological barrier-plasma membrane occurs by the following processes:
- Simple passive diffusion
- Active transport
- Carrier-mediated transport
- Facilitated diffusion
- Exchange diffusion
- Specialized process like
- Pinocytosis (cell drinking)
- Phagocytosis (cell eating)
Passive Diffusion
Drug molecules penetrate the biological barriers by passive diffusion along the concentration gradient, through aqueous channels of the cell membrane. Solute molecules are transported from higher concentration to the lower concentration. Passive diffusion of drugs across the cell membrane is a slow process, bi-directional and ceases to operate at the equilibrium phase where solute concentrations remain the same on both sides of the cell membrane. Lipid solubility, area of absorbing membrane and thickness of the membrane are the factors that govern the rate of drug absorption by passive diffusion. Cell membranes are more permeable to unionized lipid-soluble form of drugs.
Active Transport
Active transport of drugs is an energy-dependent rapid process of absorption. This biotransport of drugs occurs against a concentration gradient. Some of the examples of the drugs which are absorbed by active transport are methyldopa, L-dopa, and vitamins. Active transport is one of the basic functions of living cells. Active transport exhibits unique characteristics like selectivity, saturability and energy dependence. The movement of substances across the cell membrane is unidirectional unlike passive diffusion. Many cells prefer fructose to glucose and intestinal epithelial cells absorb sitosterol selectively in the presence of cholesterol. Saturability is a unique feature of active transport wherein active transport ceases to operate when enough concentrations of essential substances/nutrients are achieved. The secretion of H+ into the stomach, iodide trapping process by thyroid and renal absorption of glucose and amino acids are some of the examples of active transport. Cell metabolism inhibitors like dinitrophenol arrest the active transport by depriving energy for the biotransport.10
Carrier-mediated Transport
Carrier-mediated transport is a distinct type of biotransport in which the drug attaches to a cell component called “Carrier” which delivers the drug into the cell. This process does not require energy and hence known as “Facilitated Diffusion” and operates along an appropriate concentration gradient unlike active transport, for example, glucose absorption by the red blood cells and nucleoside absorption by the cells. This process is highly selective for specific conformational structure of drugs. Valinomycin is an antibiotic believed to be absorbed by facilitated diffusion.
Exchange diffusion of substances or electrolytes operates in such a way that there will not be any change in osmotic pressure of the cellular and transcellular fluids.
Pinocytosis is otherwise known as “Cell drinking”. Large molecular proteins are transported into the cell by this process, which is commonly observed in cancer cells and also in a wide variety of cells.
Factors that Modify Drug Absorption
Drugs, whether applied locally or administered systemically have to cross cell barriers to produce their action. The rate at which drugs cross biological barriers determines the time of onset of action as well as duration of action. The rate of absorption of drugs is modified by various factors, which include:
- Physicochemical characteristics of drugs
- Drug formulation factors
- Patient factors
Physicochemical Characteristics of Drugs
- Solubility
- Degree of ionisation
- Molecular size and shape
- Concentration
Solubility
Lipid-soluble drugs which are unionised are rapidly absorbed across biological barriers, e.g. thiopentone sodium, organophosphorus compounds. The extent of lipid solubility of a given drug determines the duration of action as well. Thiopentone sodium, an intravenous general anesthetic, acts rapidly because it is highly lipid soluble.
Degree of Ionisation
Most drugs are weak acids or weak bases and have one or more functional groups capable of undergoing ionisation. Biological membranes are permeable to the unionised form of drug molecule, if it is lipid soluble and relatively less permeable to ionised radicals.
The degree of ionisation of an administered drug varies with the pH of the absorbing media. Acids undergo ionisation in alkaline pH and bases ionise readily in acidic pH. Since acidic drugs remain unionised in acidic pH, these are better absorbed from gastric mucosa, e.g. aspirin and barbiturates. At physiological pH (7.4), weak acids and weak bases remain 11partly unionised and the absorption is generally proportional to the lipid solubility of the drug.
Molecular Size and Shape
If the size of the drug molecule is big, membrane permeability to such drug is rather poor. Aminoglycoside antibiotics (streptomycin) are not absorbed by oral route. The reason is streptomycin, being a large polycation molecule, fails to cross the membrane readily. Therefore, aminoglycosides are always given parenterally in systemic infections. The quaternary ammonium compounds do not cross surface barriers, for example, physostigmine, a tertiary amine is preferred to neostigmine—a quaternary ammonium compound for instillation into eye in glaucoma. However, passive diffusion of quaternary ammonium compounds may occur at high concentration.
Drug Concentration
The rate of absorption of a drug is directly proportional to concentration at the site of absorption. The higher the concentration, higher and rapid is the rate of absorption since the concentration gradient across the membrane is high.
Drug Formulation Characteristics
Drugs are available in various forms to suit the need of the patient as well as practitioner. The different types of formulations can influence the extent and rate of drug absorption. Drug solutions are readily absorbed than solid dosage forms. This is because solid drug dosage forms must undergo disintegration and dissolution as well. The disintegration rate, i.e. the time required for solid drugs to be converted into fine particles. Whereas, dissolution of the fine particles of an active drug into a solution depends on the drug substance, particle size and the pH of the absorbing site. Thus both the disintegration time and dissolution time determine the rate of absorption of the drug when given orally. In fact, this is the basis for various commercial preparations like enteric-coated tablet, sustained release/controlled release drug formulations. The rate of absorption of these preparations determines the duration of action which is solely due to drug release characteristics. In addition, co-administered solutions and mixtures do play a role in governing the rate of absorption of drugs.
Patient Factors
Many patient factors influence the rate of drug absorption, the knowledge of which is necessary for clinicians to achieve the goal of drug therapy. The factors include:
- Area of absorbing surface: The greater the area, the higher the rate of absorption.
- pH of the absorbing area: Determines the extent of ionisation of the drug.
- Bile salts pool size: Bile salts help to absorb more lipid-soluble substances from the gut.
- Presence and severity of underlying diseases: Many diseases alter the rate of absorption from gut, e.g. hypermotility disorders.
- Blood supply to the absorbing area: The drug absorption is more from highly vascularised areas, e.g. eye, buccal mucosa.
- Structure of the cell membrane: The simpler the structure of the cell membrane, better absorption.
- Body temperature: Hyperthermia usually increases the rate of absorption.
Besides the above factors, route of administration and presence of other drugs can also modify the rate of drug absorption. Intravenous route of administration can circumvent all the factors that modify drug absorption. Concurrent administration of two or more drugs do influence drug absorption. For example, in the presence of gastric antacids the absorption of tetracycline from the stomach is hindered. Conversely, ascorbic acid facilitates iron absorption.
Methods to Prolong Drug Absorption
Administration of vasoconstrictor: Simultaneous administration of a vasoconstrictor along with the drug reduces the blood supply to the site of drug administration and decreases drug absorption. However, a slow steady rate of drug absorption may be seen by this method, for example, adrenaline and lignocaine. Adrenaline being a vasoconstrictor, reduces the absorption of lignocaine from the site of injection and thus prolong the duration of action of lignocaine at the site of administration.
Application of Tourniquet: Above the site of drug injection, a tourniquet is applied to reduce the blood supply to the site, which decreases the rate of the drug absorption.
Currently, readymade drug dosage forms have been designed to achieve a steady rate of drug absorption, for example,
- Slowly disintegrating chemical complexes—procaine penicillin
- Sustained release/controlled release preparations—L-dopa
- Transdermal route of administration, e.g. ointments and patches
DRUG DISTRIBUTION
Drug distribution is the transport of a drug to its site of action, storage sites, metabolic sites and to the organs of excretion. Drug distribution determines the efficacy, duration of action, mode of metabolism and rate of excretion. Drugs are promptly distributed to heart, brain, liver, kidney or other highly perfused organs. Drugs reach less rapidly in the muscle and much more slowly in entering fat tissues. Distribution of drug in turn is dependent on two major factors—bioavailability and plasma protein-binding capacity besides other factors like drugs physicochemical characteristics, cardiac output and regional blood flow. The body compartments in which drug accumulates are the potential reservoirs of the drug.
Bioavailability
Bioavailability is the fraction/proportion of unchanged drug that reaches the systemic circulation. This is one of the important pharmacokinetic parameters of the drug that determines the intensity and duration of action.
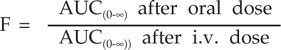

where F | = Bioavailability |
AUC | = Area under curve |
Drug formulation factors, rate of drug absorption, route of administration, dose and frequency of administration, rate of drug metabolism and excretion alter the bioavailability of drugs. Gut diseases do alter oral bioavailability of the drug. Bioavailability can be measured after the administration of the single dose or chronic administration. The concept of Bioequivalence is understood when bioavailability of various drug dosage forms of the same drug is compared.
Volume of Distribution (Vd)
Volume of distribution of a drug is the quantitative estimate of its tissue localization.
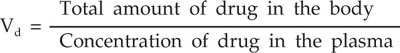
Vd is the volume of fluid in which drug appears to be distributed with a concentration equal to that of plasma. In a 70 kg man, Vd for aspirin is 11 L, digoxin 440 L and chloroquine 13,000 L. Therefore, Vd does not denote any real physiological volume. It is important for optimal drug dose response. If a drug has small Vd, it is easily dialyzable, for example, aspirin whereas drugs with high Vd like pethidine, cannot be dialysed. Volume of distribution of a drug varies with pKa, i.e. partition coefficient, protein binding, cardiac output, membrane permeability, tissue perfusion rate, age of the patient, gender and associated diseases.
Plasma Protein Binding of Drug
Large numbers of drugs on reaching circulation, bind to plasma proteins, predominantly albumin and to a lesser extent globulin. Plasma albumin exhibits high affinity for acidic drugs. Only free drug can exert pharmacological action and the protein-bound drug is pharmacologically inert. The interaction between drugs and plasma proteins is non-specific, non-selective and reversible. It dissociates rapidly whenever the free concentration of drug in plasma is reduced. Thus, the protein-bound form of the drug acts as Drug Labile Depot. Other pharmacological implications of protein binding are:
- The bound drug is not filtered at renal glomerulus.
- Extensive drug binding to plasma proteins means that the action is prolonged, e.g. warfarin—98% of an administered dose is protein bound.
- The bound form of the drug cannot diffuse through the capillary wall.
- Drugs compete each other for protein-binding sites. Consequently, displacement of one drug from binding sites by the other alters the kinetic pattern as well as pharmacodynamics of displaced drug.
Hence, to achieve the desirable therapeutic concentration of drugs in plasma in a given time, the knowledge about the drug protein-binding capacity is helpful to a clinician.
Drugs may not always be uniformly distributed—kidneys selectively accumulate mercury, cadmium and lead. Chloroquine is selectively concentrated in liver and eyes. Adipose tissue acts a storage depot for fat-soluble drugs like thiopentone and diazepam.14
Drug Reservoirs
An administered drug may be stored in cellular reservoirs like adipose tissue, bone, muscle and liver. Transcellular reservoirs like joint fluids, cerebrospinal fluid, gastrointestinal fluids, aqueous humor and endolymph also concentrate drugs.
Lipid-soluble drugs are commonly stored in neutral fat. Thiopentone, an intravenous anesthetic, is highly lipid soluble and stored in fat tissue. Bone stores tetracyclines, lead and radium. Quinacrine (Mepacrine) is stored in liver. Generally, a stored drug is released slowly from the reservoirs and hence it interferes with action of drugs.
Blood-Brain Barrier
Blood-brain barrier is a permeability barrier interposed between the circulation and the brain parenchyma. This is composed of endothelial cells, astrocytes and their basement membrane. Blood-brain barrier is less permeable to water-soluble substances. However, inflammation and disease alter the permeability of blood-brain barrier. Adrenaline, dopamine, carbidopa, quaternary ammonium compounds and various other drugs do not cross blood-brain barrier. It is believed that drugs like anticonvulsant-phenytoin alter the permeability of the blood-brain barrier.
Placental Transfer of Drugs
The maternal blood sinuses, chorionic villi, fetal membrane capillaries and trophoblast layer with mesenchymal tissue capillary endothelium structurally constitute the placental barrier. Drugs administered to pregnant women can reach fetus and produce unexpected fetotoxicities. Therefore, fetal exposure to such drugs should be avoided by taking all the precautionary measures. Morphine, antithyroid drugs, alcohol, phenytoin, anticancer drugs, oral anticoagulants and oral contraceptives cross the placental barrier and produce malformations in the fetus. Morphine can cause intrauterine fetal death due to respiratory depression. The knowledge about the placental transfer of drugs and their adverse effects on fetus is necessary for the rational use of drugs in pregnancy.
DRUG METABOLISM
The interaction between drugs and living tissues is reciprocal. Drug administration invariably results in biochemical and physiological changes in various organ systems. In turn, body brings about changes in the structure and action of drug. The metabolic transformation of the drugs inside the body is known as Biotransformation. Biotransformation of drugs occurs as a series of interdependent reactions with the product of one reaction becoming substrate for another. Drug metabolism is described to have two phases:
Pathways of Drug Biotransformation
Phase I (non-synthetic pathways of drug biotransformation)
- Oxidation
- Reduction
- Hydrolysis
Phase II: Drug conjugations (synthetic pathways of biotransformation)
- Glucuronide conjugation
- Sulfate conjugation
- Acetylation
- Methylation
- Glycine conjugation
- Glutathione conjugation
Generally, biotransformation alters the polarity, solubility, potency and results in rapid excretion of drugs. However, this is not always so. After biotransformation, a drug may become more active than the parent compound. Similarly, biotransformation is not necessary for drug elimination from the body. Many drugs are excreted as unchanged, for example, mannitol, penicillins and aminoglycosides.
Biotransformation of drugs is not the same as detoxification. The process of detoxification is involved in reducing the toxicity of the substance ingested and almost exclusively occurs in liver. A drug may become more toxic after biotransformation, for example, methanol is converted into a toxic metabolite-formaldehyde.
Sites of Drug Metabolism
Biotransformation takes place in the liver, kidney, lungs, intestine and other tissues as well.
Metabolic changes of drugs predominantly occur in the liver. However, practically in all tissues drug can undergo biotransformation. Kidneys, intestinal mucosa, intestinal microflora, plasma and lungs are also the sites of drug metabolism.
Liver is the most important organ of drug metabolism. Hepatic microsomal enzyme system extensively catalyses both phase I and phase II reactions of biotransformation. Hepatic microsomal enzyme system consists of a wide variety of group of enzymes. Lipid-soluble drugs readily gain access to hepatic microsomal enzyme system. The enzymatic activity of microsomal enzyme system of liver and other tissues are amenable to the action of drugs. Drugs can stimulate or inhibit the hepatic microsomal enzyme system. Hepatic diseases do alter the drug metabolizing capacity of the liver. Hence, in hepatic diseases, drugs with narrow safety margin are either administered at lowest effective doses or avoided.
Phase I: Non-Synthetic Pathways of Biotransformation
Oxidation
Drugs are extensively metabolized by different oxidative pathways catalysed by several distinct enzymes. The most prominent group of enzymes are mixed function oxidases of liver. These hepatic microsomal enzymes are non-specific in their function. A suprafamily of hemoproteins known as cytochrome P-450 group are the key enzymes, which take part in microsomal electron transfer system with NADPH. Several types of drug oxidases have been described—deamination, aromatic hydroxylation, N-oxidation, S-oxidation, dealkylation and desulfation. Drugs that undergo oxidation include diazepam, phenytoin, chlorpromazine, steroids, antihistamines, and adrenergic agonists.
Drugs can inhibit or stimulate the cytochrome group of enzymes and alter the rate of drug metabolism. As a result of this, various pharmacokinetic drug interactions occur. The implication and clinical significance of these interactions must be known to the physician.16
Reduction
Drugs like chloramphenicol, clonazepam undergo nitroreduction in the liver as well as other tissues. Azoreduction, dehalogenation and reduction of N-oxides and S-oxides are other known types of drug reduction pathways.
Hydrolysis
Esters, amides and glycosides are generally metabolised by hydrolysis. Succinylcholine undergoes ester hydrolysis catalysed by pseudocholinesterase present in plasma and liver.
Drug Conjugation
Drug conjugation is a systemic metabolic process involving the incorporation of endogenous radicals or groups to drug molecules or their metabolites. Conjugations take place in the liver and to a limited extent in kidneys. Drug conjugation always results in the loss of pharmacological action.
Glucuronide conjugation is catalysed by glucuronyltransferase enzyme. Morphine is usually excreted as morphine glucuronide. Many drugs including paracetamol and chloramphenicol are excreted as glucuronides.
Drugs which are primary amines generally undergo acetylation catalysed by acetyltransferase in liver, for example, isoniazid.
Adrenaline, histamine, phenols and thiols are the agents for “methylation” whereas acetylsalicylicacid undergoes glycine conjugation. Ethacrynic acid is excreted as glutathione conjugate.
Fate of Drugs after Metabolism
Biotransformation brings changes in pharmacokinetic as well as pharmacodynamics of drugs, which may be beneficial and often harmful. Lipid-soluble drugs are converted into more water-soluble substances. Non-polar compounds will be converted to polar and thus facilitates elimination of drugs. Further, drug metabolism may result in:
- Change in the Nature of ActionBiotransformation can change the nature of action of the drug, for example,

- Increased Potency → ActivationExamples:
- Diazepam → Oxazepam
- Imipramine → Desmethylimipramine
- Talampicillin → Ampicillin
When drug potency is increased after biotransformation due to the release of active metabolites, the concept is regarded to as activation of drugs. The parent compound, which is inactive or less active than its metabolite, is known as prodrug, for example, dipivefrine and talampicillin. - Increased toxicityMany drugs become more toxic after metabolism. Methanol on biotransformation releases formaldehyde, which is neurotoxic. The clinical implication of these metabolic changes, which are observed with drugs, must be known to all. One of the metabolites of cyclophosphamide, an anticancer drug, causes hemorrhagic cystitis.
Variation in Drug Metabolism
There are great individual variations in drug metabolism. Frequently, these variations are under genetic control. Physiological factors like age, gender, pregnancy, presence of hepatic disease, environmental factors and stimulation and inhibition of drug metabolising enzymes by drugs cause variation in the rate and types of drug metabolism.
Enzyme Induction and Enzyme Inhibition
Many enzymatic metabolic reactions are augmented by drug themselves. This phenomenon is known as Enzyme Induction. Drugs that stimulate enzymes are called Enzyme Inducers for example, phenobarbitone, rifampicin, phenytoin and alcohol. Certain drugs stimulate their own metabolism by stimulating the metabolising enzyme activity.
The enzyme induction caused by drugs may at times result in clinically significant variation in the efficacy of drug therapy. When a female patient is on oral contraceptive therapy, if rifampicin is prescribed concurrently, oral contraceptive measure may fail resulting in pregnancy. The reason is rifampicin being an enzyme inducer enhances the metabolism of hormonal contraceptive eventually leading to reduced efficacy of the contraceptive.
Conversely, there are drugs that can inhibit hepatic drug metabolising enzyme system, for example, cimetidine, ciprofloxacin and erythromycin. As a result, this causes variation in the efficacy of concurrently administered drug.
Genetic Variation in Drug Metabolism
Genetically determined polymodal variation in biotransformation of drugs is common. The metabolism of succinylcholine, isoniazid, primaquine and other drugs show genetically determined variation in their metabolism. Often, this may turn out to be fatal. Therefore, it is important to study the genetic polymorphism in drug metabolism to avoid complications of drug treatment.
Succinylcholine is a skeletal muscle relaxant that undergoes ester hydrolysis in plasma and liver catalysed by pseudocholinesterase. The people of Greece, Israel, Portugal, 2% British and North Africans produce atypical pseudocholinesterase which fails to hydrolyse succinylcholine. Administration of succinylcholine to these individuals results in succinylcholine apnea, which may lead to paralysis of respiratory muscles and diaphragm.
Isoniazid is a first line antitubercular agent which is metabolised by acetylation. Rate of isoniazid acetylation is genetically determined. The human population can be grouped 18into two, based on the rate of isoniazid acetylation—‘slow’ acetylators and ‘rapid’ acetylators. Of late, ‘medium’ acetylators group is being added. It is important to realize that doses of isoniazid have to be titrated depending upon the rate of acetylation in a given patient. This is necessary to avoid any variation in the efficacy and toxicity in a patient.
Genetic factors not only alter the metabolism but also toxicity profile of the drug. Primaquine is an antimalarial innocuous drug. However, if a patient is deficient of glucose-6 phosphate dehydrogenase enzyme, primaquine can cause death causing extensive hemolysis. Pertinently, human genomic research is trying to unleash the details of the relationship between gene and drug kinetics. Till then, clinicians must focus well and be constantly vigilant to avoid drug-induced hazards based on genetic defects in particular.
First Pass Metabolism
The rate of drug metabolism may vary when administrated by different routes. Orally administered drugs can be degraded during their transit before reaching systemic circulation. Drugs like catecholamines, insulin and lignocaine cannot be given by oral route. These drugs are completely destroyed in the gastrointestinal tract and while passing through liver. Drug metabolism that occurs before reaching systemic circulation is described as first pass metabolism.
Lignocaine, propranolol, insulin and catecholamines are a few examples of drugs that undergo extensive first pass metabolism. For this reason, lignocaine is not given orally as an antiarrhythmic agent. However, propranolol can be given by oral route at recommended doses to produce intended action. In fact, on first pass metabolism, an administered drug may be converted into an active metabolite and a rapid action seen. Thus, potency and efficacy of a drug remains same, after first pass metabolism for prodrugs like enalapril.
DRUG EXCRETION
The elimination of drugs and their metabolites occur by several routes. Kidneys, intestine, lungs, and biliary system account for most part of drug excretion. However, renal excretion is by far the major route and to a lesser extent, sweat, salivary and mammary glands in lactating mother also contribute for the elimination of drugs. Generally, water-soluble compounds are more readily eliminated.
Clearance
The time required for complete elimination of administered dose of a drug is called clearance. The total body clearance of a drug is the direct expression of the body's ability to eliminate drug. In general, the rate of biotransformation to eliminate drug is fairly constant. Nevertheless, drug clearance is dependent upon blood flow to the organs of excretion, protein binding, plasma concentration, hepatic and renal diseases. The principle of clearance is similar to that of renal physiology. Thus, reduction in drug dosages and less frequent administration may be suitable for most drugs in the conditions of renal disease. Therefore, the knowledge about drug clearance is useful in predicting the response in a given condition. The duration of action and plasma concentration are conveniently expressed in terms of half-time.
19
Many drugs like penicillin, probenecid, digoxin and thiazide diuretics are excreted by active renal tubular secretion. When these drugs are given together, they compete for the renal secretory pathways. Thus, kinetics and dynamics of the affected drug may change. For example, thiazide diuretics compete with uric acid for tubular excretion and uric acid is reabsorbed. The elevated uric acid levels in plasma can precipitate gouty arthritis. Therefore, thiazide diuretics are contraindicated in gout. Physicians need to be aware of drug interactions that occur at the sites of drug excretion.
Plasma Half-life (t1/2)
Plasma half-life of a drug is the time taken for the elimination of half of the plasma concentration of the administered dose. The plasma half-life is an index of plasma concentration and duration of action, which is useful particularly for the optimization of drug dose regimen for chronic treatment. The rate at which drug leaves the body is dependent on volume of distribution and clearance. Hence, plasma half-life of drug expresses the relationship between volume of distribution and dose. Plasma half-life of the drug is an estimate to determine appropriate dosing interval and good indicator of the time required to reach plasma steady state concentration of the drug. As with other pharmacokinetic parameters, plasma half-life of drugs varies with factors like severity of disease, age, protein binding, tissue binding and the rate of absorption.
Forced Alkaline Diuresis
The rate of drug excretion can be enhanced by promoting their ionization in plasma, since ionized radicals are rapidly excreted. Acidic drugs undergo ionization in alkaline pH and vice versa. Hence, if acidic drugs like barbiturates and salicylates, when ingested in large quantities, their excretion can be enhanced by alkalinization of plasma to reduce toxicity. Further, a diuretic can be used along with sodium bicarbonate to increase the rate of drug elimination. This is known as forced alkaline diuresis. Forced alkaline diuresis is commonly employed in acute salicylate and barbiturate poisoning conditions.
Enterohepatic Circulation
It means that a drug may be excreted by the liver cells into the bile and eliminated through intestine. From intestine, drug may be absorbed back to reach liver. In this way, drug may be shunted from liver to intestine and intestine to liver. This is referred to as enterohepatic circulation. Penicillins, copper, steroid hormones, procainamide, sulfonamides, chloramphenicol, tetracyclines and chlorpromazine are the few examples of drugs that undergo enterohepatic circulation.
Excretion by other Methods
Pulmonary Excretion
Volatile general anesthetics and paraldehyde are the drugs that are excreted through lungs. Only 2% of the ingested alcohol is excreted through this route since major part of alcohol is metabolised extensively.20
Excretion through Hair and Nails
Griseofulvin, an antifungal antibiotic, gets deposited in hair follicles and nails. In fact, this has a clinical advantage. Griseofulvin is the drug of choice to treat fungal infections of hair follicles and nail. Arsenic and mercury are also excreted through hair and nails.
Excretion through Sweat
Quinine, mepacrine, arsenic and mercury are excreted in sweat. Mepacrine produces yellowish discoloration of skin as it gets excreted in the sweat.
Excretion through Saliva
Metronidazole, an antiamoebic drug, gets excreted through saliva producing metallic taste in the mouth. Other examples of drugs that appear in saliva are atropine, clonidine, barbiturates and sulfonamides.
Drugs that are Distributed into Milk
Many drugs are excreted through milk, for example, isoniazid, chloramphenicol, diazepam, oral anticoagulants and antithyroid drugs. When a nursing mother is on drug therapy with agents, which are known to appear in milk, she should be advised not to feed the baby. Otherwise the baby may be exposed to alarming drug toxicity. If the nursing mother on diazepam treatment feeds her baby, the baby may be sedated unduly. Therefore, knowledge about drug excretion in milk is useful for clinicians while prescribing drugs for lactating mothers.
First/Zero Order Kinetics
First Order Kinetics
When the rate of drug administration equals the rate of elimination, the concentration of the drug in plasma remains steady. Drug dose and steady state plasma concentration are directly proportional, i.e. doubling the dose must lead to a two-fold increase in plasma concentration ideally. For many drugs, an exponential time course follows before they disappear from plasma. The relationship between dose and rate of excretion is said to be linear; hence, it is known as first order or linear kinetics.
Zero Order Kinetics
When the drug dose and plasma steady state concentration relationship is non-linear, non-exponential, it is known as zero order kinetics or non-linear or saturation kinetics. Phenytoin, alcohol and heparin are the few examples of drugs that obey zero order kinetics. Drug saturation kinetics bear important clinical consequences. There is likelihood of gradual accumulation of drugs and difficulty to achieve a steady concentration in plasma because of variation in kinetics from individual to individual. This necessitates plasma concentration monitoring to ensure safety. This method is known as therapeutic drug monitoring.21
PHARMACODYNAMICS
DEFINITION AND INTRODUCTION
Pharmacodynamics is that branch of pharmacology, which deals with action and mechanism of action of drugs. Drugs produce their action interacting with various cellular mechanisms and events. It is important to recognize that drugs cannot create new functions in the body nor restore the lost functions of any system, which happened due to structural defects. Indeed, drugs can only impart changes in biochemical and physiological processes in the living body to produce their effects.
The action of drugs may be produced by two major ways:
- Specific action involving a known type of receptors.
- Actions produced by nonreceptor mechanisms. For example, alcohol-induced action, which does not involve receptors.
DRUG RECEPTORS
Definition of Receptor
A receptor is a macromolecular biophysical unit with which the drug interacts that leads to a sequence of events culminating as a characteristic action of the drug.
A drug, which exhibits both affinity and efficacy on receptor interaction, is called the ‘agonist’ of that particular type of receptor. Affinity of the drug is the tendency to form drug-receptor complex. Efficacy or intrinsic activity of a drug is defined as the ability to induce physico-chemical events, which occur subsequent to the formation of drug receptor complex that results in drug action. Currently, this is referred to as receptor signal transduction pathway.
A drug which has affinity for the receptor but lacks efficacy is known as ‘antagonist’. For example, atropine blocks the muscarinic receptor. It binds with muscarinic receptor but has no intrinsic activity.
An agonist with full receptor occupancy fails to produce maximum effect is known as ‘partial agonist’, for example; buprenorphine is an µ opioid receptor partial agonist. Whereas an agonist on receptor occupation produces opposite effects to that of classical agonist of the same receptor type is known as ‘inverse agonist’, for example, ²-carbolines on benzodiazepine receptors.
Many therapeutically useful drugs act either as agonists or antagonists of the known types of receptors. Currently, different types of drug receptors have been studied thoroughly and characterised well by their structure and function. A drug binds to receptors by ionic, 22hydrogen, covalent bonds and van der Waals’ force. If a drug establishes covalent bond with a receptor, the interaction is said to be stable and irreversible.
Further, drugs bind to enzymes, carrier molecules and ion channels to produce their action. This evinces that no drug is completely specific in producing actions. However, it is certain that receptors form the sensing elements in the system of chemical communications that co-ordinates drug action as well as cellular function. The precise chain of events occurs following drug receptor interaction which depends upon the particular receptor and the cell. The receptors may be directly linked to ion channels, enzyme kinases, nuclear substances or coupled with G-protein system for signal transduction.
FACTORS THAT MODIFY DRUG ACTION
Biological variation in the action of drugs is a natural phenomenon. Many factors cause variation in drug action, which are as follows:
- Physical factors: Body weight and body surface
- Physiological factors: Age, sex, temperature, pregnancy, posture, mental attitude and patient's compliance
- Pathological factors: Presence of disease and severity of the disease
- Pharmacological factors: Structure of the drug, dose bio-availability, presence of other drugs, route administration, time and frequency of administration and tolerance
- Genetical factors: Species variation and idiosyncrasy
- Environmental factors: ‘CO’-rich air pollution
Age
Age is an important factor in determining the response to a given drug. The variability of drug action to an extent is age dependent. At large, the effects of age on the type of quantitative drug response are inseparable. The individuals at the extremes of lifespan are often unusually sensitive to drugs. Apparently, this is due to changes in rates of absorption, distribution, biotransformation and excretion.
In children, liver is ill equipped with enzymes to metabolise drugs. The renal excretion of drugs is also depressed. Further, the volume of blood flowing through the kidney per unit time is smaller in the infant in proportion to the total body water content. Therefore, drug action tends to be intense or prolonged or both. Consequently, effective drug levels persist for longer periods. For example, chloramphenicol is more toxic to infant because the rate of metabolism of this antibiotic is slow and hence produces ‘gray baby syndrome’. Hence, chloramphenicol is contraindicated in neonates.
With aging, there is a normal decline in the integrity of the various organs and systems (homeostenosis). In the elderly people end-organ sensitivity, blood flow to organs and ability to handle drugs are quite different when compared to an adult patient. Hence, geriatric (aged) patients are likely to respond to drugs in quantitatively different ways than the average adult does. For example: it is not unusual to see paradoxical excitement to a sedative hypnotic drug diazepam in an elderly patient. Geriatric patients are readily vulnerable to digoxin toxicity. To avoid complications, drug dose regimens need to be altered for both paediatric and geriatric patients.23
Gender
The variation in drug action attributable to gender appears to be of little consequence. However, on the basis of weight, women may need smaller doses of drugs than men. Obviously, sex hormones produce different actions when given to individuals of opposite sex. Estrogens produce feminisation of male and masculinisation trends are seen with androgens in female. Barring these natural differences in drug actions based on gender are infrequent and inconsequential.
Structure of the Drug
The structure of the drug is intimately associated with the action. Any minor/major modification in the structure may alter the kinetic as well as dynamic characteristics of the drug. The study of structure-activity relationship of drugs has been beneficial to produce various congeners of established official drugs. Often, these congeners ensure more potency, efficacy and safety. The knowledge about the structure-activity relationship is of immense value to explore the nature of drug receptor interaction. As a result, it is made possible to synthesize various agonist and antagonist to that particular receptor.
It is not surprising that hardly few changes have been observed in the structure of noradrenaline, adrenaline and dopamine. However, the adrenoceptor activity of these drugs differs substantially. Adrenaline acts on both ± and ² receptors, whereas dopamine receptor agonistic activity is entirely different.
Dose
Dose of a drug is defined as the quantity of the drug that is administered to produce a predictable action in a given clinical condition. Obviously, the dose employed in any clinical circumstances must produce maximal therapeutic benefit and minimal or no toxicities. The action produced by a drug in general is directly proportional to the dose administered. The dose of a drug required to produce 50% of the maximal effect of that drug is described as “drug potency”. The potency of drug is determined by drug affinity and coupling reaction with the receptor. Nonetheless, the selection of the drug is based on its efficacy but not potency. The clinical efficacy of a drug depends on the maximal effect it produces in a patient. Further, the drug efficacy may depend on the route of administration, absorption, distribution and clearance from the site of action. Drug potency is largely determined by the dose to be given for a particular patient. Hence, there is no reason to believe that the more potent drug is clinically preferred as it is being stressed in drug advertisement.
Route of Administration
Route of administration is an important factor in altering the onset, intensity and often the nature of action. Many drugs fail to reach systemic circulation as they undergo complete inactivation when given by mouth, for example, insulin and other protein drugs. A change in the route of drug administration of a drug may result in the production of different pharmacological actions as seen with magnesium sulphate. Orally administered magnesium 24sulphate produces purgative action. When given by intravenous route, it acts as central nervous system depressant and on local application induces anaesthetic effect. Thus, often the selection of route of administration is an important therapeutic exercise at least in part, to attain clinical efficacy from the drug treatment.
Presence of Other Drugs
Concurrent administration of two or more drugs is a common practice and often a clinical necessity. Multiple drug therapy leads to either beneficial or at times harmful drug interactions. It is well recognised that combination drug therapy results in two types of drug—drug interactions synergism and antagonism involving both pharmacokinetic and pharmacodynamic mechanisms.
DRUG SYNERGISM
Drug synergism is defined as the positive summation of drug effects. In synergism the combined effect of two drugs is greater than the algebraic sum of their individual effects. Often, the term drug potentiation is used as interchangeable word with synergism of drug actions. Therapeutically applied drug combinations for synergistic effects are plenty. Trimethoprim + sulphamethoxazole as antifolate drugs, diuretic with other antihypertensives hydrochlorothiazide + atenolol, organic nitrites with ² adrenergic blockers as antianginal drugs, antitubercular—3 or 4 or 5 drug regimens—isoniazid, rifampicin, ethambutol, pyrazinamide, streptomycin and co-administration of enzyme inhibitors which inhibit the metabolism of drugs are a few examples for achievable therapeutic synergy for the benefit of the patient.
DRUG ANTAGONISM
The negative summation of drug effects is drug antagonism. This is to say that the combined effect of two or more drugs is less than the algebraic sum of the individual effects. Drug antagonism is of different types. Mainly four types have been described:
- Physical antagonism
- Chemical antagonism
- Physiological or functional antagonism
- Pharmacological antagonism:
- Competitive
- Non-competitive
Physical Antagonism
This type of drug antagonism is mainly due to physical properties of the agents. One drug is inactivated in direct proportion to an extent of opposing physical properties of the other, for example, heparin is a negatively charged macromolecule binds to positively charged proteins like protamine. Thus formed protamine-heparin complex is pharmacologically inactive. For this reason, protamine is used as an antidote in heparin overdose. Nevertheless, protamine is a strong base and heparin is acidic. These properties of drugs also contribute for antagonistic interactions.25
Chemical Antagonism
When the chemical reaction that occurs between two drugs form the basis for drug interaction with negative outcome, it is known as chemical antagonism. For example, sodium bicarbonate and hydrochloric acid being acid and alkali neutralise the gastric acid and useful in acid peptic diseases.
Functional or Physiological Antagonism
This is observed when two endogenous substances acting at different sites produce mutually antagonistic effects. Histamine is a powerful bronchoconstrictor and adrenaline counteracts the action of histamine on bronchial smooth muscle. This interaction occurs as a physiological regulatory tissue mechanism hence the name—functional antagonism.
Pharmacological Antagonism
The antagonism observed at receptor level between two drugs on administration of therapeutic/recommended doses is known as pharmacologic antagonism. The cardinal feature of pharmacologic antagonism is as a rule, both the agonist and antagonist act on the same receptor. In many clinical conditions pharmacologic antagonism form the basis for the administration of specific antidotes to save the life of patient. For example, in acute opioid poisoning—naloxone a specific opioid antagonist and atropine in organophosphorous poisoning.
Drug receptor antagonism is of two types:
- Competitive drug antagonism
- Non-competitive drug antagonism
Competitive drug antagonism is also known as surmountable or reversible antagonism. In this type of antagonism, the action of an antagonist can be reversed by increasing the dose of agonist by many folds. The agonist administered at high doses displaces the antagonist from the receptor and produces its action on the receptor. Acetylcholine and atropine interaction at muscarinic receptor sites is said to be ‘competitive antagonism’. Atropine action on muscarinic receptor can be reversed by increasing the concentration of acetylcholine at the immediate vicinity of receptors.
In non-competitive (unsurmountable, irreversible) drug antagonism, it is not possible to regain the effect of agonist by increasing its dose. For example, the action of glutamate on its receptor cannot be regained once the receptor is occupied by a non-competitive antagonist memantadine. The interaction between noradrenaline and phenoxybenzamine at stage two establishing covalent bond is also referred to as irreversible antagonism.
DRUG TOLERANCE
Drug tolerance is a state of decreased responsibility to the administered dose of a drug and to achieve the same intensity/degree of response doses needs to be increased. Drug tolerance may involve both kinetic and dynamic mechanisms or alone either. Generally, decreased responsiveness to the given dose of a drug due to variation in absorption is referred to as ‘pseudotolerance’. Whereas, tolerance due to various changes that occur at the target sites of drugs is known as ‘pharmacodynamic tolerance’. Primarily, the mechanisms that are responsible for the development of drug tolerance include increase in the rate of 26metabolism, rapid excretion, desensitisation of receptors, which may be associated with downregulation of receptors and genetic trends also contribute.
The development of drug tolerance may be acute or chronic. The acute tolerance to drug action is otherwise called ‘tachyphylaxis’. Tachyphylaxis or acute tolerance is defined as a state of reduced responsibility to the administered dose of a drug given within short time intervals. The exact mechanisms that give rise to tachyphylaxis remain uncertain. The development of tachyphylaxis seem to involve:
- Enhanced drug disposition due to increased rate in biotransformation
- Rapid elimination
- Fast adaption of drug site of action to the state of reduced sensitivity—exhaustion of stored neurotransmitters, downregulation of receptors and desensitisation of receptor.
The common examples of drugs that produce tachyphylaxis include ephedrine, tyramine, 5-hydroxytriptamine and prazosin. Histamine, nitroglycerin, and amphetamine are also known to provoke tachyphylaxis. The clinical significance of tachyphylaxis is of paramount importance, especially in chronic drug therapy. The management of high blood pressure is a perennial exercise; drug used for this must be free from the phenomenon of the development of tolerance to their action.
Cross-tolerance is seen when a patient develops tolerance to a drug and remains tolerant to other drugs that belong to the same group. For example, morphine tolerant may show tolerance to the action of pethidine.
Drug distribution patterns also account for the development of tolerance. Thiopental, an intravenous general anaesthetic, is a highly lipid-soluble drug. On administration, it reaches the brain and acts for 11–20 minutes. This ultra short action is due to its redistribution. After a lapse of brief period of time, the active drug diffuses from brain and reaches adipose tissue where it is stored. The administered dose of thiopental is very much inside the body but unavailable at the site of action. Hence, this is known as ‘drug redistribution tolerance’.
DRUG TOXICITY
Drugs produce many actions. All the drug actions may not be required to achieve therapeutic benefit. The unwanted actions of a drug seen along with the desired main pharmacological actions are commonly described as ‘adverse drug reactions’. Adverse drug reaction includes (a) side effects and (b) toxic effect. A side effect appears at therapeutic doses, which is not wanted and inseparable from the main actions of drugs. For example, morphine is used for analgesic purposes but causes constipation and respiratory depression as side effects. Dryness of mouth produced by many drugs is also a classic example for drug-induced side effect.
All drugs do produce toxicities when administered at supramaximal doses. Toxic effects of drugs are always due to high abnormal dose administration. Drug-induced toxicities may be predictable or unpredictable. Predictable toxicity is often an extension of therapeutic action of drug and related to dose administered. The unpredictable drug toxicity is not always related to dose and often unintentional. Drug allergic manifestations are regarded as unpredictable toxicities when observed at first. Drugs can cause structural toxicities like hepatotoxicity, haematological, renal, pulmonary, gastrointestinal and neurotoxicities besides mutagenicity, carcinogenicity and teratogenicity.27
Iatrogenic Disease
A drug- or physician-induced disease is called ‘iatrogenic disease’. The etiology of iatrogenic disease is not difficult to describe. However, physician's awareness on drug-induced toxicities can minimise this adversity. Glucocorticoid-induced Cushing's syndrome and digitalis intoxication are the noted examples for iatrogenic diseases.
Carcinogenicity
The occurrence of cancer inducing agents is abundant in nature. Carcinogenicity has been observed with estrogen and some anticancer drugs like cyclophosphamide. Many environmental chemicals including nicotine cause cancer. Alcohol is regarded to be a procarcinogenic.
Mutagenicity
An agent that causes mutation is known as mutagen. Drugs like dacarbazine, 5-fluorouracil and cytarabine are known to be mutagens. Drug-induced mutagenicity may be one of the reasons for the development of cancer.
Teratogenicity
Drugs that induce malformation of foetus and intrauterine death are regarded as ‘teratogens’. The first trimester of pregnancy is the most vulnerable period for drug-induced foetal deformation, especially at the time of organogenesis. Many drugs are teratogenic, for example, phenytoin, alcohol, warfarin, sodium valproate and thalidomide. Phenytoin causes ‘foetal hydantoin syndrome’, alcohol produces ‘foetal alcohol syndrome’, whereas sodium valproate induces neural tube defect called ‘spina bifida’.
Cumulative Toxicity
Cumulative toxicity produced by drugs is invariably due to frequent administration. Drugs, which have long plasma half-lives like digoxin, boric acid, and emetine, are the common examples known to cause cumulative toxicity. Drugs accumulate in the body in great proportions to produce cumulative toxicity. Optimisation and individualization of drug dose regimen can readily avoid cumulative toxicity.
MEASURE OF DRUG SAFETY
Therapeutic Index
To produce a desired effect, information about the safe use of drugs needs to be furnished with an assessment of margin of drug safety. This measure ought to relate the dose of the drug required to produce an intended action and an undesired action. This is commonly referred to as ‘therapeutic index’. It is the ratio between median lethal dose to median effective dose for animals.
Therapeutic index = LD50/ED50
LD50 : Lethal Dose in 50% of the animals used
ED50 : Effective Dose in 50% of the animals
28
If the therapeutic index of a drug is one, that drug is said to be equally toxic. Ideally, therapeutic index should be more than one and preferably a high TD50 to ensure more safety.
The Placebo
The word meaning of this Latin term placebo is ‘I shall please’ or ‘I please’ or ‘I may please’! A placebo resembles the active drug in all respects by appearance. Placebo is a dummy medicament that resembles the active drug in its shape, size, dose, colour and method of administration. Placebos are used as vehicles to cure the ailments by suggestions (not by true actions by any means). Lactose, gelatin and fructose are generally used as placebo medicaments. Tonics are placebos in a true sense except when prescribed for extremely debilitated patients. Not all subjects react to placebo administration. The percentage of placebo reactors is about 40%. Placebo can produce side effects like perspiration, nausea, headache and anorexia. Placebo is mainly employed in the clinical evaluation of drug development studies to avoid human bias. However, deliberate use of placebo is a confession of failure on the part of clinician.
DEVELOPMENT OF NEW DRUGS
The development of a new drug involves time, finance and risk. The procedure is complex (Fig. 1.1) and expensive and extremely arduous. Here the research studies need to be ethically stringent and well designed. Mainly there are two approaches to design a new drug development programme viz.
- Rational approach in which chemicals are synthesized in the light of detailed knowledge of biological process and tested for efficacy and safety.
- Random approach aims at conducting a valley of tests for substances and by chance useful activity is found.
Nevertheless, both the approaches must carry out a sequence of experimentation using two/more species of animals. The preclinical testing generally identifies the characteristic activities of a test drug. Both the primary and secondary animal screening of drugs depict activity and selectivity of drugs under study, besides assessing the safety profile. The animal study of the test drug provides evidences for efficacy in relation to safety at the doses employed. These data paves the way for human testing. A well-designed ethical investigation of drugs in human beings for their rational use is called ‘clinical trial’. The clinical trial is a teamwork that comprises healthy normal human volunteers, clinicians, pharmacologists, statisticians and patients. The chief aim of the clinical trial is to reduce a number of variable factors of drug treatment while assuring safety and efficacy.
The clinical trials must be well executed to avoid bias on the part of investigators and patients as well. To avoid human bias, placebo-controlled studies have been the procedures followed in the clinical trials. The clinical trial involves four phases. Phase I study in human volunteers to declare that the test drug is fit for human use or not. In Phase II dosage requirement, dynamic and kinetic profile of the drugs is to be determined with the active participation of limited number of patients. Whereas the long-term safety and efficacy of the test drug is evaluated in phase III employing more number of patients. Phase III study critically evaluates the overall therapeutic value of the test drug. Adverse drug reactions monitoring will invariably continue till the drug is used. This includes the phase IV study in clinical trial also known as “post-marketing surveillance”.29
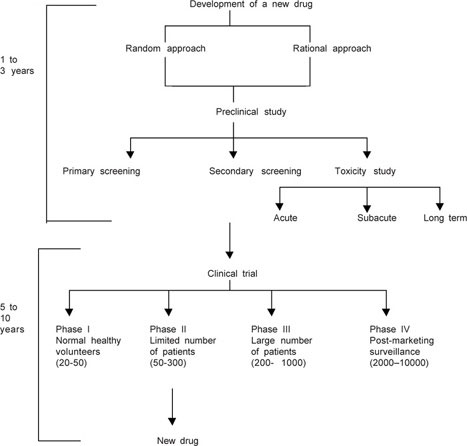
Phase IV examines marketing authorization given to drug developer and drugs may be withdrawn from the market if the agents found them to be more toxic. Post-marketing surveillance also explores new combinations and new methods of administration by gathering pharmacoepidemiological data.