



INTRODUCTION
Congenital lid anomalies are a heterogeneous group of disorders. Broadly they may be classified into malformations and malpositions of the eyelids. Some conditions may be innocuous, while others may have serious visual consequences, and need prompt attention.
DEVELOPMENT OF EYELIDS
The eyelids develop from lid folds that first appear in the surface ectoderm overlying the optic vesicle in the 16 mm stage of the embryo, and grow till the 32 mm stage. Between the 32 to 37 mm stages of the embryo, the upper and lower lid folds fuse together. They begin to separate at the fifth month of gestational age, starting at the nasal side, and the separation is completed by the sixth month.
CRYPTOPHTHALMOS
Etiology
Signs and Symptoms
Cryptophthalmos presents with three grades of severity.
- Complete cryptophthalmos is usually associated with other developmental anomalies, and often bilaterally symme-trical. A smooth skin passes from the forehead to the cheek. Eyebrows and lashes are usually absent (Figure 1A).
- Partial cryptophthalmos shows partially formed eyelids and a disorganized anterior segment. The lids are often colobomatous. Facial skin fuses with the medial aspect of the globe (Figure 1B).
- Abortive cryptophthalmos has a formed globe, with partial absence of the eyelids in some parts; symblepharon is seen at these areas. Cornea is covered by keratinised and stratified epithelium (Figure 1C).
Associated ophthalmic features include microphthalmos, anterior segment anomalies such as small anterior chamber, absent trabecular meshwork, subluxated lens, posterior segment anomalies like uveal coloboma, adnexal abnormalities like dermoids, supernumerary brow and absent lacrimal and accessory glands.
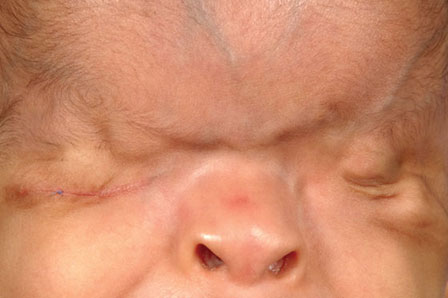

Figure 1B: Partial cryptophthalmos. There is a disorganized anterior segment and partial formation of eyelids

Figure 1C: Abortive cryptophthalmos, with incomplete formation of the nasal end of the eyelids, with symblepharon formation
Cryptophthalmos may also have associated non-ophthalmic abnormalities, including cleft lip or palate, hernia, aplasia of kidneys, malformed genitalia, meningo-myelocele, and mental retardation.
Differential Diagnosis
Complete cryptophthalmos has a characteristic appearance that is diagnostic. Incomplete cryptophthalmos needs to be differentiated from isolated lid colobomas. Causes of acquired symblepharon are to be differentiated fron abortive cryptophthalmos.
Investigation
Ultrasound B scan and computed tomography may help to assess the condition of the globe.
A thorough systemic evaluation is required to detect any associated anomalies.
Management
The management is surgical, with staged reconstruction of eyelids and the ocular surface. The visual outcome is poor.
LID COLOBOMAS
Etiology
Lid colobomas form due to delay in fusion of the mesodermal components of the fronto-nasal and maxillary processes of the face.
Signs and Symptoms
A coloboma is a partial or full thickness absence of the eyelid, which may be triangular, quadrilateral, W-shaped or irregular. The edges are rounded and covered with conjunctiva. The partial thickness coloboma may show the lid margin structures such as lashes. The upper lid colobomas are usually in the middle and the nasal third of the lid. The lower lid 6colobomas are often situated temporally. If situated at the medial end of the lower lid, they are associated with abnormalities of the lacrimal drainage system.
The cornea may be visible through the coloboma even when the lid is closed, and show exposure keratopathy on slit-lap examination (Figure 2A).
Differential Diagnosis
Congenital colobomas need to be differentiated from acquired coloboma caused by trauma.
A coloboma may be isolated, or be a part of the Goldenhar syndrome.
Investigation
The child requires systemic evaluation to rule out other congenital abnoamalities.
Management
The urgency of surgical repair of a coloboma depends on the severity of exposure caused by it. Any coloboma larger than one-third the horizontal extent of the eyelid poses danger to the cornea, and must have early surgical correction. Any smaller one may undergo elective correction, and have conservative management with lubricants, moist chambers or bandage contact lenses. The edges of the coloboma should be manually pulled together to assess the actual size of the coloboma, to plan appropriate surgery. Smaller ones may have direct closure. Larger ones may need more extensive reconstruction. When using a lid-sharing technique, the risk of amblyopia in a young child should be remembered (Figures 2A and B).7

Figure 2A: Clinical photograph of patient with right eye lateral canthal ectopia, congenital ptosis, and left eye upper lid coloboma with corneal scarring due to exposure

Goldenhar Syndrome
Lid coloboma is a common association with Goldenhar syndrome, in addition to corneal or epibulbar dermoids, and pre-auricular skin tags and external ear deformities (Figures 3A and B).8

Figures 3A and B: Goldenhar syndrome with left eye upper lid coloboma and pre-auricular and external nasal skin tags
MICROBLEPHARON AND ABLEPHARON
Introduction and Signs & Symptoms
A microblepharon is a rare vertical foreshortening of the eyelid. The upper lid is more commonly affected. Apparent ablepharon may be a severe microblepharon, and one should look carefully for rudimentary eyelids. It may be associated with clinical anophthalmos. It is associated with lagophthalmos and exposure keratopathy. There may be additional ocular and systemic abnormalities (Figure 4).
Investigation
Evaluation should be performed for addditonal systemic and ocular abnormalities. In a microphthalmic eye, assessment for visual potential is required.

Figure 4: Patient with right eye euryblepharon, left eye clinical anophthalmos with upper and lower lid microblepharon. The eyelids are short vertically as well as horizontally
Differential Diagnosis
Microblepharon and ablepharon should be differentiated from lid colobomas.
Management
The surgical management plan depends on the visual potential and the specific features. Correction may require rotation flaps from the cheek, lid sharing procedures, pedicle flaps or skin grafts.
ANKYLOBLEPHARON
Ankyloblepharon Filiforme Adnatum
Signs and Symptoms
The condition shows fine extensile cords attaching upper and lower lids, which may be single or multiple. The cords decrease the palpebral fissure height and reduce lid excursion. It has been reported n association with trisomy 18.
This may be associated with ectodermal dysplasia and cleft lip or cleft palate – this is the autosomal dominant Hay-Well's syndrome.
Treatment
The cord is simply severed, and the epithelial tags involute.
Congenital Ankyloblepharon
Introduction
Congenital ankyloblepharon is caused by developmental arrest leading to growth aberration at either canthus, inner or outer.11
Signs and Symptoms
It is caused by partial fusion of the upper and lower lid, and results in horizontal foreshortening of the palpebral fissure. The commoner external variety is at the lateral canthus, causing an appearance of pseudoexotropia. The internal ankyloblepharon at the inner canthus causes pseudo-esotropia.
Differential Diagnosis
Pseudo-eso- or exotropia must be differentiated from true strabismus.
Treatment
Ankyloblepharon requires surgical separation of the eyelids.
MEDIAL CANTHAL ECTOPIA
Introduction
Medial canthal ectopia is caused by developmental arrest in the second month of intrauterine life.
Signs and Symptoms
The medial canthal tendon is displaced inferiorly along with surrounding canthal structures. It attaches to the junction of the medial wall of the orbit and the inferior orbital rim. There may be associated nasal cleft deformities and nasolacrimal duct obstruction (Figure 5).
Differential Diagnosis

Figure 5: Clinical photograph of patient with right eye corneal dermoid, medial canthal ectopia both eyes and small colobomatous lid notching in left eye. The medial canthus is displaced downwards, and is associated with congenital nasolacrimal duct obstruction. The patient has undergone surgical correction of cleft lip and palate previously
Investigation
The child should be assessed for nas-lacrimal duct obstruction.
Treatment
The condition requires surgical supraplacement of the medial canthal tendon: a Z-plasty or a Y-V plasty may be used. The associated nasolacrimal duct obstruction should also be corrected.
EPICANTHUS
Introduction and Signs & Symptoms
Epicanthus is a semi-lunar fold of skin at the medial canthus, with its concavity directed outward. The epicanthal fold obscures the view of the medial canthal angle, the caruncle and plica. An epicanthus may be isolated or be an associated feature in a patient with congenital ptosis or Blepharophimosis syndrome. There are several varieties of epicanthus.
- Epicanthus tarsalis extends from the upper lid
- Epicanthus inversus extends from the lower lid (associated with Blepharophimosis-ptosis-epicanthus inversus syndrome—BPES)
- Epicanthus palpebralis extends from the upper lid to the lower lid
- Epicanthus superciliaris extends from below the eye-brow.Epicanthus may give an appearance of pseudoesotropia (Figures 6A to C).
Differential Diagnosis
Any epicanthus should be examined to rule out associated BPES.

Figure 6A: Patient with right eye congenital ptosis, with epicanthus tarsalis both eyes. A fold of skin extends from the upper lid to the lower lid at the medial canthus

Figure 6B: Patient with bilateral congenital ptosis and bilateral epicanthus inversus. A semilunar fold of skin extends from the upper lid to the lower lid

Figure 6C: Epicanthus palpebralis, with a semilunar fold of skin extending on both upper and lower lids. The epicanthal fold obscures the medial canthal structures and gives an appearance of pseudostrabismus
Treatment
Epicanthus is corrected by Mustarde double Z-plasty, modified Y-V plasty or C-U plasty.
CENTURION SYNDROME
Introduction and Signs & Symptoms
The patient of centurion syndrome has a history of epiphora since childhood, which worsens at puberty. Syringing of the nasolacrimal duct is patent. The syndrome derives its name from the resemblance of the patients to the Roman centurions in their helmets. The medial canthal tendon is taut and attached anteriorly, causing medial stand-off of the lower lid and punctum. The lacrimal punctum is displaced forward out of the lacrimal lake, causing epiphora. The facial appearance is typical, with a sharp medial canthus, long horizontal palpebral fissure and square eyebrows (Figure 7).
Differential Diagnosis

Figure 7: Patient with centurion syndrome, with sharp medial canthal angles, square eyebrows, and elongated horizontal palpebral aperture. The medial canthal tendon is prominent and attached anteriorly, not allowing proper apposition of the lacrimal punctum to the tear meniscus
Investigation
Lacrimal syringing will be patent, but dye disappearance will be delayed.
Treatment
The treatment is surgical, anterior medial canthal tendon release, with medial canthoplasty or punctoplasty.
DISTICHIASIS
Introduction and Signs & Symptoms
Distichiasis is an extra row of eyelashes behind the normal ciliary margin at the sites of meibomian glands. It may involve the whole length of the lid or only a segment. The distichiatic lashes rubbing on the ocular surface can induce epiphora or even keratopathy. Congenital distichiasis is associated with lower limb lymphedema; the gene responsible is FOXC 2 (Figure 8).16

Figure 8: Incomplete row of eyelashes growing on the medial aspect of the upper lid; the lashes are at the sites of meibomian gland orifices
Differential Diagnosis
Distichiasis is to be differentiated from other conditions with abnormal positions or abnormal directions of eyelashes, such as trichiasis, epiblepharon and entropion.
Investigation
Instillation of fluorescein in the eye will help to detect early keratopathy.
Treatment
If the patient is asymptomatic, observation alone suffices. A small number of lashes may be treated by electrolysis. In extensive distichiasis, the lid can be split along the grey line and cryo applied to the posterior lamella at the roots of the distichiatic eyelashes.17
EPIBLEPHARON
Introduction and Signs & Symptoms
Epiblepharon comprises a horizontal redundant lid fold of skin and orbicularis in the lower lid, causing vertical orientation of the eyelashes. There may be coexistent entropion. The eyelashes rubbing on the cornea cause epiphora, and may cause keratopathy. It is commoner in Mongolian features. It may be associated with inferior oblique weakness. Epiblepharon often resolves spontaneously by 3 years of age (Figures 9A and B).
Differential Diagnosis
The condition is to be differentiated from congenital entropion and distichiasis.
Investigation
Instillation of fluorescein dye will help to detect any keratopathy. Careful examination will rule out associated inferior oblique weakness.
Treatment
Epiblepharon may be treated with lubricants in the expectation of spontaneous resolution. If the cornea shows keratopathy, surgical intervention is required.


Figure 9B: Clinical photograph showing redundant horizontal fold of lower lid skin and orbicularis, with vertical orientation of eyelashes
The redundant skin and orbicularis oculi muscle is excised in the form of a spindle; usually 1–3 mm of excision would suffice. In severe cases everting sutures may be used in addition.
CONGENITAL ENTROPION
Introduction
Congenital entropion affects the lower lid, and results from inadequate development of the lower lid retractors. This causes lower lid malpositions similar to that in senile entropion. The entropion is often accompanied by epiblepharon and epicanthus (Figure 10).
Signs and Symptoms
In congenital entropion the lower lid rolls inwards, and the eyelashes rub on the ocular surface. The patient may be symptomatic, with epiphora and keratopathy.19

Figure 10: Clinical photograph of patient with bilateral congenital ptosis, with anti-mongoloid slant of palpebral fissures, with lower lid entropion. Left eye shows a failed corneal graft and lower lid external hordeolum
Differential Diagnosis
Congenital entropion must be distinguished from epiblepharon and distichiasis.
Management
Surgical correction is required if the patient develops keratopathy. The procedure of choice is reattachment of the lower lid retractors to the tarsus.
TARSAL KINK SYNDROME
Introduction and Signs & Symptoms
Tarsal kink syndrome is a rare entity with horizontal fold along the entire horizontal length of the tarsus. Tarsal kink results in entropion of the upper lid; in the severest cases, the child presents within a few weeks of age, and the lid margin is not visible. The patient may have absent lid crease, blepharospasm, lid edema and keratopathy. The patient commonly presents with corneal ulcer in early infancy (Figures 11A to C).20

Figures 11A and B: Patient with bilateral tarsal kink and bilateral corneal ulcers. The upper lids show severe entropion, with obscuration of lid margin and absent lid crease
Differential Diagnosis
Tarsal kink syndrome with corneal infiltrate should be differentiated from patients presenting with other corneal ulcers.21

Figure 11C: The everted eyelid shows a crease in the tarsal plate along the entire horizontal length
Management
Surgical correction of the tarsal kink entails full-thickness blepharotomy the entire length of eyelid with marginal rotation. Due to the very early age of presentation and the associated corneal ulceration and scarring, amblyopia therapy is crucial after correction of the entropion (Figure 11D).

CONGENITAL ECTROPION
Introduction and Signs & Symptoms
Congenital ectropion of the lower lid is rarely isolated; it is more likely to be a part of a syndrome such as Kohn Romano syndrome (Figures 12A and B). Congenital ectropion of the lower lid often has vertical shortage of skin. Severe ectropion will cause epiphora and exposure, and the tarsal conjunctiva may become keratinized.
Lamellar ichthyosis is a systemic condition which may result in cicatricial ectropion of upper and lower lid at birth.
Occasionally there is an isolated eversion of the upper lid, for which the etiology is postulated as birth trauma causing vascular stasis and congestion, leading to lid eversion (Figure 13).
Differential Diagnosis
The ectropion due to shortage of skin should be differentiated from euryblepharon, which also causes lateral ectropion of the lower lid.
Management
Congenital ectropion requires full-thickness skin grafting; the preferred donor site is the retroauricular area.

Figure 12A: Clinical photograph of patient with bilateral congenital ptosis, blepharophimosis, telecanthus and lower lid ectropion

Figure 12B: The lower lid shows severe ectropion, vertical shortening of the anterior lamella, keratinization and pigmentation of the conjunctiva. The cornea shows exposure keratopathy

In lamellar ichthyosis, it may be difficult to identify any area of healthy skin as a potential donor site. Skin emollients should also be used liberally.
In the isolated upper lid eversion, one may escape surgery, and get by only with marginal traction suture to straighten the lid. If the ectropion persists, one may need to excise a part 24of the prolapsed conjunctiva to allow correction of the lid eversion.
EURYBLEPHARON
Introduction and Signs & Symptoms
Euryblepharon presents with bilateral symmetrical enlargement of the horizontal palpebral apertures, with vertical shortage of eyelid skin, elongated lid margins, and downward and lateral displacement of outer canthi. The horizontal palpebral fissure length is increased to approximately 35 mm from the average of 28–30 mm. There is lateral ectropion, lagophthalmos and reduced blink rate with exposure keratopathy. The exposure and reduced blink rate may cause epiphora (Figures 14A to C).
Differential Diagnosis
Euryblepharon should be distinguished from the congenital ectropion which has vertical shortage of skin, but not an elongated lid margin.
Other causes of epiphora in childhood should be differentiated from euryblepharon.

Figure 14A: Clinical photograph of patient with bilateral upper and lower lid lateral ectropion, with excess horizontal length

Figure 14B: Photograph of the same patient after surgical correction of the euryblepharon. The corneas are scarred due to exposure

Figure 14C: Photograph of patient with mild degree of euryblepharon, with lateral ectropion of lower lid alone
Management
Mild degrees may be managed conservatively, with lubricants. Moderate degrees will require tarsorrhaphy to protect the cornea. A lateral canthoplasty with shortening of the upper and lower lids laterally may be beneficial. The severest forms may require staged correction with augmentation of the posterior lamella and anterior skin graft.26
CONGENITAL LID RETRACTION
Introduction and Signs & Symptoms
Congenital retraction of the upper lid may be unilateral or bilateral. A unilateral lid retraction may simulate contralateral ptosis. The condition is non-progressive. Other systemic features are usually normal.
Differential Diagnosis
Congenital lid retraction must be differentiated from other causes of lid retraction such as thyroid abnormalities, and Marcus Gunn phenomenon. Children developing eyelid retraction must be investigated for intracranial space-occupying lesions, hydrocephalus and postencephalitic sequelae (Figures 15A and B).
There has been a case report of accessory slip of levator palpebrae superioris causing lid retraction.
Investigation
Thyroid function tests may rule out endocrine-related lid retraction.
Management
Lid retraction may be treated by lid lengthening procedures.
CONGENITAL PTOSIS
Introduction and Signs & Symptoms
Congenital ptosis is the commonest of the congenital lid anomalies. The upper lid is at a lower level than normal, and the condition may be unilateral or bilateral. The affected eye may have limited elevation. A quick assessment of the severity of congenital ptosis is possible by looking at the red reflex of the pupil through distant direct ophthalmoscopy, and recording how much of the pupil is obscured by the lid.27


Other indicators of the severity of the ptosis are over-action of the frontalis muscles and chin elevation on inspection of the face. A greater palpebral fissure height in down gaze is seen in the unilateral ptosis, since the defective levator palpebrae muscle is also deficient in ability to relax. A faint upper lid crease indicates weak levator action. Marcus Gunn phenomenon, i.e. synkinetic elevation of the ptotic lid with jaw movement should be looked for. Amblyopia may develop due to visual deprivation, refractive error or associated strabismus (Figures 16A and B).28


Differential Diagnosis
Careful examination will rule out other causes of ptosis in the young.
Myasthenia gravis may rarely occur in a child. Variable ptosis, involvement of extraocular muscles and fatigability indicate myasthenic ptosis. The diagnosis may be confirmed by edrophonium test.
Chronic progressive external ophthalmoplegia may have an onset in the first decade of life, and show associated limitation of ocular movements.
Congenital ocular fibrosis presents with ptosis along with strabismus. The ocular movements are severely restricted.29
Ptosis may be associated with weakness of the third cranial nerve.
Investigation
The child with ptosis requires careful cycloplegic refraction. Ice test and edrophomnium test help to rule out ocular myasthenia.
Management
Severe ptosis with high risk of amblyopia should be corrected early in life. Milder disease is corrected as the child reaches the school-going age. Severe ptosis with poor levator action is treated by a tarso-frontal sling; the material of the sling may be biogenic such as autogenous or preserved fascia lata or bovine pericardium, or synthetic such as mersilene, silicone, PTFE or poly-propylene. Any associated strabismus requires correction before correction of the ptosis.
Ptosis with associated Marcus Gunn phenomenon needs special consideration. Disinsertion of the levator aponeurosis from the tarsal attachment bilaterally with bilateral tarsofrontal sling has been advocated. An alternative is to disinsert the levator of the affected side alone, with bilateral sling surgery.
After correction of the ptosis, refractive error should be reassessed and correction provided. Amblyopia therapy should be instituted if required.
BLEPHAROPHIMOSIS SYNDROME
Introduction and Signs & Symptoms
Blepharophimosis is characterized by a short horizontal palpebral fissure, 18–22 mm as opposed to the average of 28–30 mm. Blepharoptosis is severe, with poor levator function, absent lid crease and characteristic head posture with chin elevation and over-action of the frontalis. Epicanthus inversus shows a semilunar fold of skin at the medial canthus, extending from the lower lid to the upper lid. Telecanthus is an increased distance between the orbits. The inter-medial canthal distance is greater than half the inter-pupillary distance. The tetrad of Blepharophimosis, blepharoptosis, epicanthus inversus and telecanthus together comprise the Kohn Romano syndrome. Blepharophimosis syndrome may, in addition, have inferiorly slanted palpebral apertures and lateral ectropion.
The Blepharophimosis, Ptosis, Epicanthus inversus Syndrome (BPES syndrome) has been identified to be autosomal dominant. There are two types of this syndrome: BPES Type I is associated with infertility, while BPES Type II is not. The gene involved in BPES Type I is FOXL2.
The Callahan classification of blepharophimosis is based on the particular combination of clinical features at presentation:
Blepharophimosis type I: Blepharoptosis, blepharophimosis, epicanthus inversus, telecanthus.
Blepharophimosis type II: Blepharophimosis and blepharoptosis, without epicanthus and normally spaced orbits
Blepharophimosis type III: Blepharophimosis and blepharop-tosis, with shortage of skin and palpebral apertures slanted inferiorly, with wide spaced orbits.
Differential Diagnosis
Management
Blepharophimosis syndrome requires staged repair. The epicanthus is corrected first, by Mustarde's double Z-plasty or by modified Y-V plasty. Any lateral ectropion causing lagophthalmos and exposure is corrected by lateral canthoplasty, and skin grafting if necessary. The ptosis is corrected by a tarsofrontal sling surgery (Figures 17A to C).

Figure 17A: Blepharophimosis syndrome type 1, with ptosis, blepharophimosis, telecanthus and epicanthus inversus


OTHER SYNDROMES
There are several syndromes which are associated with lid anomalies. A few of the important ones are listed, with the responsible gene and clinical features mentioned in brief.
Prader Willi Syndrome
- Gene responsible—SNRP small nuclear ribonucleoprotein polypeptide)
- Clinical features—almond shaped palpebral fissures, obesity, hypogonadism, mental retardation
Noonan Syndrome
- Gene responsible PTPN 11 (protein tyrosinase phosphatase non-receptor type).
- Clinical features—hypertelorism, down slanting palpebral fissures, webbing neck, pulmonary stenosis, pectus excavatum.
Rubinstein Taybi Syndrome
- Gene responsible—CRE binding protein
- Clinical features—heavy arched eyebrows, down slanting palpebral fissures, broad thumbs and toes, mental retardation.
Fraser Syndrome
- Gene responsible—FRAS1 (ECM protein)
- Clinical features—cryptophthalmos, renal agenesis, syndactyly, laryngeal stenosis.
Apert Syndrome
- Gene responsible—FGFR2
- Clinical features—severe craniostenosis, syndactyly, hypertelorism, proptosis, strabismus.
Crouzon Syndrome and Related Syndromes (Pfeiffer and Jackson Weis syndrome)
- Gene responsible—FGFR2-FGFR
- Clinical features—craniostenosis, limb anomalies (absent in JWS, PS), hypertelorism, proptosis, orbital asymmetry.
Saethre-Chotzen Syndrome
- Gene responsible—TWIST
- Clinical features—craniostenosis, limb and ear anomalies, ptosis.
Waardenburg Syndrome
- Gene responsible—PAX3 (WS1), MITF (WS2)
- Clinical features—iris heterochromia, deafness, white forelock, dystopia canthorum (WS1)
Treacher Collins Syndrome
- Gene responsible—TCOF1
- Clinical features—first branchial arch syndrome, down slanting palpebral fissures, eyelid colobomas.
Coffin Lowry Syndrome
- Gene responsible—RPS6KA3
- Clinical features—X linked mental retardation syndrome, hypertelorism, down slanting palpebral fissures.
Stickler
- Gene responsible—COL2A1, COL11A1, COL11A2
- Clinical features—midfacial hypoplasia, cleft palate, hearing loss, spondyloepiphyseal dysplasia, proptosis.
BIBLIOGRAPHY
- Bernardini FP, Kersten RC, de Conciliis C, Devoto MH. Unilateral microblepharon. Ophthal Plast Reconstr Surg. 2004 Nov; 20(6:)467–9.
- Dawodu OA. Total eversion of the upper eyelids in a newborn. Niger Postgrad Med J. 2001 Sep; 8(3:)145–7.
- DN, Ferrell RE, Meisler DM. Lymphedema-distichiasis syndrome and FOXC2 gene mutation. Am J Ophthalmol. 2002 Oct; 134(4:)592–6.
- Dollfus H, Verloes A. Dysmorphology and the orbital region: a practical clinical approach. Surv Ophthalmol. 2004 Nov-Dec; 49(6:)547–61.
- Egier D, Orton R, Allen L, Siu VM. Bilateral complete isolated cryptophthalmos: a case report. Ophthalmic Genet. 2005 Dec; 26(4:)185–9.
- Jain S, Atkinson AJ, Hopkisson B. Ankyloblepharon filiforme adnatum. Br J Ophthalmol. 1997 Aug; 81(8:)708.
- Kao YS, Lin CH, Fang RH. Epicanthoplasty with modified Y-V advancement procedure. Plast Reconstr Surg. 1998 Nov; 102(6:)1835–41.
- Naik MN, Honavar SG, Bhaduri A, Linberg JV. Congenital horizontal tarsal kink: a single-center experience with 6 cases. Ophthalmology. 2007 Aug; 114(8:)1564–8.
- Nouby G. Congenital upper eyelid coloboma and cryptophthalmos. Ophthal Plast Reconstr Surg. 2002 Sep; 18(5:)373–7.
- Oculoplastic, Orbital and Reconstructive Surgery. Edition 1. Albert Hornblass Ed. Williams and Wilkins, Baltimore. 1988,
- Ptosis, Edition 3. C Beard. CV Mosby Company, St Louis. 1981,
- Samlaska CP. Congenital lymphedema and distichiasis. Pediatr Dermatol. 2002 Mar-Apr; 19(2:)139–41.
- Sires BS. Congenital horizontal tarsal kink: clinical characteristics from a large series. Ophthal Plast Reconstr Surg. 1999 Sep; 15(5:)355–9.
- Spierer A, Bourla N. Primary congenital upper eyelid retraction in infants and children. Ophthal Plast Reconstr Surg. 2004 May; 20(3:)246–8.
- Sujatha Y, Sathish S, Stewart WB. Centurion syndrome and its surgical management. Ophthal Plast Reconstr Surg. 1999 Jul; 15(4:)243–4.
- Sullivan TJ, Welham RA, Collin JR. Centurion syndrome. Idiopathic anterior displacement of the medial canthus. Ophthalmology. 1993 Mar; 100(3:)328–33.
- Traboulsi EI, Al-Khayer K, Matsumoto M, Kimak MA, Crowe S, Wilson SE, Finegold
- Tüysüz B, Ilikkan B, Vural M, Perk Y. Ankyloblepharon filiforme adnatum (AFA) associated with trisomy 18. Turk J Pediatr. 2002 Oct-Dec; 44(4:)360–2.
- Wylen EL, Brown MS, Rich LS, Hesse RJ. Supernumerary orbital muscle in congenital eyelid retraction. Ophthal Plast Reconstr Surg. 2001 Mar; 17(2:)120–2.