

HISTORICAL BACKGROUND
The virus was visualized under the electron microscope for the first time in 1970 by Dane and Almeida and their respective colleagues, who described the infectious form of the virus or Dane particle, and its nucleocapsid core, respectively. This was soon followed by the characterization of the virus genome, the proteins it encodes as well as their function. The setting-up of diagnostic assays for various markers of infection at the same time facilitated the definition of the serological profiles that characterize acute and chronic infection with the virus.
Outbreaks of hepatitis among shipyard workers in Bremen and inmates of an asylum for the mentally insane in Merzig were described towards the end of the 19th century, following vaccination against smallpox. The condition was referred to at the time as serum hepatitis and the link with its causative agent, namely HBV, was not made until the mid-1960s. Studies, performed by Krugman and colleagues at the Willowbrook State School for the mentally handicapped children during this time established that hepatitis A virus and HBV were the causative agents for infectious and serum hepatitis respectively. The presence of Australia antigen in the sera of patients with leukemia had been described a few years earlier, but its connection with HBV and hepatitis B surface antigen (HBsAg) in particular, as it is now known, did not become apparent until later studies performed by the teams of Blumberg and Prince. These studies set the groundwork for the subsequent serological tests for the diagnosis of HBV and allowed detailed investigations into the epidemiological and virological aspects of infection, and connection of the virus with the development of hepatocellular carcinoma (HCC) in high prevalence areas of the world.
The molecular biology of the virus was unravelled in the years that followed with the development of genetic engineering techniques that allowed the cloning of the viral genome, study of the function of its proteins and revealed the fascinating mechanism of its replication strategy. The polymerase chain reaction allowed the speedy amplification and sequencing of virus isolates that led to the identification of its genotypes, quasispecies and virus mutants, using bioinformatic tools.
A landmark in a historical context was the development and introduction of a plasma-derived prophylactic vaccine in the early 1980s, following its extensive 2evaluation by Smuzness and colleagues in chimpanzees and the gay community in the United States of America. This vaccine was soon replaced by its recombinant counterpart, and more than 25 years on, the vaccine has proved extremely efficacious at reducing HBV prevalence rates in many countries where the virus is endemic. Almost concurrently, interferons were introduced for the treatment of chronic HBV carriers in an attempt to stem the progression to cirrhosis and HCC. Nowadays, apart from interferon which most treatment guidelines recommend that it be used for up to a year, longer term treatment is possible with nucleos(t)ide analogs that inhibit DNA synthesis and the reverse transcriptase (rt)/DNA polymerase enzyme of the virus.
TAXONOMY AND CLASSIFICATION
Hepatitis B virus is the prototype member of the hepadnaviridae, a family of hepatotropic DNA viruses, which are divided into two genera. Other than HBV, the orthohepadnavirus genus includes members that infect rodents such as woodchucks (woodchuck hepatitis virus, WHV) and ground squirrels (ground squirrel hepatitis virus). These viruses have about 70% sequence homology with HBV but do not appear to infect man or other primates. In contrast, HBV is transmissible to chimpanzees, whilst HBV-like isolates have been obtained from primates such as gibbons, gorillas, orangutans, woolly monkeys, as well as chimpanzees. More distantly related viruses and with almost no sequence homology to HBV are those of the genus avihepadnavirus which infect birds such as ducks (duck hepatitis B virus, DHBV), herons, storks and geese. In the absence of a cell culture system for the propagation of HBV, the chimpanzee, woodchuck and duck animal models have proved invaluable over the years, in the study of the natural history of infection, molecular biology of the virus, and the testing for efficacy of vaccines and antiviral drugs.
VIRION STRUCTURE
Under the electron microscope virus preparations from serum appear pleomorphic. Three types of particle are evident—the infectious virions or Dane particles which measure about 42 nm in diameter and have an electron dense core, and two types of subviral particles devoid of nucleic acid that consist entirely of HBsAg. The latter are the 22 nm spheres and filaments which outnumber the virions by 100-10000-fold. The infectious virion has an outer envelope which consists of HBsAg in a lipid bilayer that fully enwraps the nucleocapsid core of the virus (Fig. 1.1). The latter consists of the hepatitis B core protein (HBcAg) and encloses the DNA genome of the virus together with its rt/DNA polymerase.
GENOME ORGANIZATION/TRANSCRIPTION
The DNA genome of the virus is a relaxed circle in shape, partially double stranded and of a total length of around 3200 base pairs. This compact nature of the genome makes HBV the smallest known DNA virus pathogenic to man.


Fig. 1.1: Structure of the hepatitis B virion. A. Electron micrograph of HBV purified from plasma showing the infectious Dane, and the spherical and filamentous subviral particles. B. Structure of the virion and its components.(Reprinted from the Encyclopedia of Virology, Third Edition, Karayiannis and Thomas “Hepatitis B Virus: General Features”, p350-60, 2008)
The genome is partially double stranded since the positive strand is incomplete, whilst the negative strand is nicked. The genome contains 4 wholly or partially overlapping open reading frames (ORFs) (Fig. 1.2) and what is more, all its regulatory elements such as enhancers, promoters, encapsidation and replication signals lie within these ORFs.
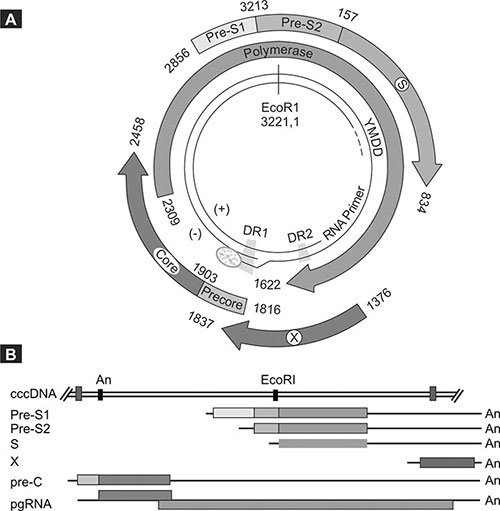
Fig. 1.2: Genome organization of the virus showing the various open reading frames (ORFs) and some of the regulatory elements (A). The transcripts encoding for the various viral proteins are co-terminal but of various lengths as shown (B). The color coding is the same for the ORFs in both panels.(Reprinted from the Encyclopedia of Virology, Third Edition, Karayiannis and Thomas “Hepatitis B Virus: General Features”, p350-60, 2008)
4The S ORF encodes for the HBsAg proteins, the P ORF for the rt/DNA polymerase and the X ORF for the X protein which is thought to be a transactivator of viral and cellular genes with a role in hepatocarcinogenesis. Finally the C ORF encodes for the HBcAg or core protein which forms the nucleocapsid of the virus. As explained below, an additional nonstructural protein is produced during replication, and this protein is known as hepatitis B e-antigen (HBeAg).
The viral proteins are translated from the appropriate mRNAs, the synthesis (transcription) of which is carried out in the nucleus by the host RNA polymerase II. Following infection as described later, the viral genome is delivered to the nucleus where it undergoes repair. The incomplete strand is completed and the ends of each of the two strands are ligated, leading to the formation of a completely double stranded molecule known as covalent closed circular DNA (cccDNA), capable of operating as a mini-chromosome and a prerequisite step for successful gene transcription. Each gene of HBV has one or more promoters regulating its activity and these promoters are in turn regulated by one or both viral enhancer elements (En1 and En2), located upstream of the basal core promoter (Fig. 1.2). Moreover, transcription is dependent to a large extent upon liver-enriched transcription factors. All of the transcripts terminate at a common polyadenylation signal and consist of the subgenomic mRNAs that encode for the surface and X proteins, and two longer than genome length ones (3.5 kb) known as the PreC-mRNA and the pregenomic RNA (pgRNA). The synthesis of both of these RNAs is under the control of the basal core promoter located in the X ORF. Due to the circular nature of the cccDNA template, these mRNAs have a terminal redundancy so that the sequences at the beginning of the RNA (5’) are repeated at the end (3’). They arise as a result of the RNA polymerase by-passing the polyadenylation signal during its first passage but not doing so during the second.
The Pre-C mRNA encodes for HBeAg, whilst the pgRNA does so for both the core and polymerase proteins. Moreover, the latter also serves as the template for reverse transcription into DNA during replication of the virus.
ENVELOPE PROTEINS
The S ORF encodes three envelope glycoproteins produced by differential initiation of translation at each of three in-frame initiation codons, which are known as the large (L), middle (M) and small (S) HBsAgs. The proteins are in fact translated from three separate transcripts and are coterminal (Fig. 1.2). Thus the C-terminus of both the L and M proteins is shared in its entirety with the complete amino acid sequence of the more abundant S protein, and is referred to as the S domain. The M protein has at its N-terminus an additional 55 amino acids encoded by the Pre-S2 region, whilst the L protein includes apart from the Pre-S2 domain another 125 amino acids at its N-terminus encoded by the Pre-S1 region (Fig. 1.2). All three proteins are glycosylated whilst the L and S proteins may be present in an 5unglycosylated form in particles also. They are type II transmembrane proteins weaving in and out of the lipid bilayer membrane of the endoplasmic reticulum (ER), where they are inserted during their synthesis. The proteins form multimers stabilized through disulfide bridges through cysteine residues in the S domain. All three of them form the outer envelope of the Dane particle, but the ratios vary with the S protein representing around 70% of the HBsAg content of the virion. The main virus neutralizing epitope is contained in the S domain and is often referred to as the Major Hydrophilic Region or the “a” determinant. The extent of this region remains undefined, but it is thought to lie between amino acid positions 100–160 of the S domain and to constitute a conformational cluster of epitopes, through the formation of disulfide bridges between cysteine residues. Virus neutralizing epitopes also exist in the Pre-S1 and Pre-S2 domains. The Pre-S1 protein is thought to contain between amino acid positions 23–47 the region responsible for virus interaction with the hepatocyte receptor. Moreover, it is thought that that Pre-S1 protein is also involved in binding the nucleocapsids that are formed within the cytoplasm. This entails that the Pre-S1 domain of a fraction of the L protein be maintained on the cytosolic side of the ER. Finally, the L protein undergoes myristylation of its N-terminus, a process which is not required for efficient virus assembly but is required for infectivity and membrane association.
CORE PROTEIN
The C ORF contains two in-frame initiation codons and can, therefore, encode for two separate translation products from two different mRNAs. Initiation of translation from the first initiation codon borne by the PreC-mRNA results in synthesis of the precore protein consisting of 29 amino acids from the precore domain whilst the remainder is identical with the core protein. This protein constitutes the precursor of HBeAg, a soluble nonstructural protein and marker of active virus replication. A signal peptide at the N-terminus of the protein tethers it to the ER membrane, so that the rest is localized within the ER lumen. Action by a signal peptidase within the lumen results in the removal of the signal peptide (first 19 amino acids), whilst further proteolytic processing at the C-terminus of the protein leads to the loss of an additional 23 amino acids from that end. What remains of the protein is the HBeAg. This is thought to have a tolerogenic effect on the immune response to the virus, which characterizes the first phase of chronic infection, particularly in those exposed to the virus in early childhood (immune tolerant phase). Loss of HBeAg and development of the corresponding antibody (anti-HBe) is one of the primary goals of antiviral therapy.
The nucleocapsid or core protein (HBcAg) is synthesized through usage of the second initiation codon present in the pgRNA. The core protein has the capacity to dimerize through disulfide bonding, followed by self assembly into the icosahydral structure that constitutes the viral nucleocapsid. The latter consists of 6240 copies of the core protein or 120 dimers, with surface projections as revealed by X-ray crystallography. The tips of these projections bare a major B-cell epitope that is recognized by anti-HBc antibodies.
X PROTEIN
The X ORF overlaps with the En1 and basal core promoter regions. The function and role of the encoded X protein in the life cycle of the virus remains largely unknown. The protein is essential for infectivity in vivo and antibodies against the protein are present in patients’ sera. In vitro studies have established that the protein operates as a multifunctional regulator modulating gene transcription, protein degradation, apoptosis and signaling pathways. More precisely, it activates a broad variety of different promoter elements and interferes with signaling cascades upstream from the transcription complex. Transcription factors that may be affected include AP1, NF-kB, SP1 and oct-1. It affects the expression of a variety of genes involved in the cell cycle, proliferation or apoptosis, can localize in mitochondrial membranes inducing oxidative stress and may interfere with DNA repair. All of these effects may contribute to the accumulation of critical mutations in the hepatocyte DNA and may explain the hepatocarcinogenic potential of the protein. Its potential role in malignant transformation has been clearly demonstrated in transgenic mice following over expression of the protein in these animals.
RT/DNA POLYMERASE
The P ORF covers almost ¾ of the viral genome and encodes for the polymerase of the virus. As already mentioned, the pgRNA encodes for both the core and polymerase proteins of the virus. The P ORF initiation codon lies in the distal part of the core gene, and thus the two ORFs overlap, but they are not in frame. Preferential binding of ribosomes to the 5’ end of the pgRNA leads to translation of the C ORF and accumulation of the core protein. This is not surprising as multiple copies of the core protein are required for capsid formation. Translation of the P ORF is, therefore, is less efficient compared to that of the C ORF. At some stage though, polymerase synthesis occurs followed by binding of the protein to the 5’ end of its own pgRNA. This event arrests further synthesis of the core protein and initiates the process of encapsidation.
The polymerase has three functional domains which starting at the N-terminus include the terminal protein involved in DNA priming, the rt/DNA polymerase domain and the RNAse H domain that occupies the C-terminus of the protein and is involved in pgRNA degradation. There is also a spacer region of unknown function situated between the terminal protein and the rt domain. Molecular studies have shown that this region can be deleted without a detrimental effect on replication competence. The rt domain contains the characteristic YMDD motif (Tyrosine-methionine-aspartic acid-aspartic acid) mutations which have been 7associated with resistance to nucleoside analogs such as lamivudine. Host cell factors including chaperones from the heat shock protein family, two of which are Hsp70 and Hsp90, are thought to be instrumental in aiding encapsidation, stabilization and activation of the polymerase.
REPLICATION STRATEGY
As stated already the pgRNA has a terminal redundancy which duplicates the nucleotide sequence at its 5’ end of this RNA up to the polyadenylation signal, and encompasses the direct repeat 1 (DR1) region to the beginning of the C ORF. The intervening sequences which happen to overlap with the precore region form a secondary structure known as the epsilon (ε) or encapsidation signal. It is formed through intramolecular base pairing of palindromic sequences that lead to the formation of a structure consisting of a lower stem, an upper stem, a side bulge and an apical loop (Fig. 1.3). Once synthesized, the polymerase engages through recognition of its conformational structure. Concurrently with this event, as its name implies, it effects the encapsidation of the pgRNA/polymerase complex into the nucleocapsid particle, where all subsequent steps in virus nucleic acid synthesis take place. Although the PreC-mRNA is only slightly longer than the pgRNA, only the latter is encapsidated.
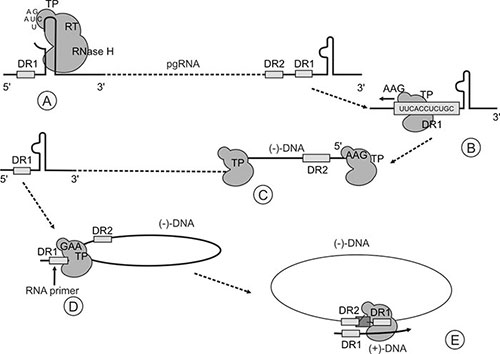
Fig. 1.3: Replication strategy of the virus. A. Primer synthesis; B. Translocation and binding to DR1; C. Synthesis of the (–)-DNA strand; D. Preservation of the RNA primer fragment from RNase H degradation. E. (+)-DNA strand synthesis.(Reprinted from the Encyclopedia of Virology, Third Edition, Karayiannis and Thomas “Hepatitis B Virus: General Features”, p350-60, 2008)
Of the duplicated elements in the pgRNA mentioned above, two, namely the at the 5’ and the DR1 at the 3’ ends of the RNA are crucial elements in the successful replication of the viral nucleic acid.
The interaction between the polymerase and # sets in motion the events that initiate reverse transcription of the pgRNA to the negative (–)-DNA strand of the relaxed circular HBV-DNA. The side bulge of the # structure (Fig. 1.3) serves as the template for the synthesis of a 3-4 nucleotide long DNA primer. The synthesis of the primer is primed by the N-terminal domain of the polymerase encompassing the terminal protein. The primer is covalently attached to the polymerase through a phosphodiester linkage between dGTP and the hydroxyl group of a tyrosine residue at position 63 of the terminal protein. This event involves the # structure at the 5’ end of the pgRNA, and is then followed by the translocation of the polymerase-primer complex to the 3’, where it hybridizes with the DR1 region with which it is homologous. Elongation of the bound primer, and thus synthesis of the (–)-DNA strand, occurs by reverse transcription of the pgRNA template. In the process, the pgRNA template is degraded by the action of the RNase H domain of the polymerase. However, the RNase H degradation stops short of the 5’ of the pgRNA, thus preserving an RNA oligomer consisting of 11-16 ribonucleotides that encompass the DR1 sequence at this end, still attached to the cap structure.
A second translocation event then takes place which allows the DR1 RNA primer to bind to DR2 located at the 5’ end of the newly synthesized (–)-DNA strand. This translocation is assisted by the circularization of the (–)-DNA strand which is brought about by long-distance interactions between segments of this strand which are not directly part of the donor and acceptor elements involved in the translocation events. In any rate, as the polymerase is covalently attached to the 5’ end of the (–)-DNA strand, the latter is effectively circularized during its synthesis. In spite of this, the long-distance interactions are then necessary to bring into juxtaposition the DRI and DR2 sites. The RNA primer containing DR1 hybridizes with DR2 with which it is homologous, initiating (+)-DNA strand synthesis and using the (–)-DNA strand as template. The polymerase extends the primer a short distance towards the end of the 5’ end of the (–)-DNA strand and then a template exchange occurs that allows the (+)-DNA strand synthesis to proceed along the 3’ end of the complete (–)-DNA strand, effectively circularizing the genome. As already mentioned, DNA synthesis occurs within the nucleocapsid and this is facilitated through pores allowing entry of nucleotides. Once the maturing nucleocapsid is enveloped by budding through the endoplasmic reticulum membrane, the nucleotide pool within the capsid cannot be replenished, hence the incomplete nature of the (+)-DNA strand.
VIRUS LIFE CYCLE
The life cycle of the virus begins with its attachment to the appropriate hepatocyte receptor (Fig. 1.4), which still remains unknown. The virion is internalized probably by endocytosis, the envelope is removed and the nucleocapsids are then released in the cytosol.

Fig. 1.4: Diagrammatic representation of the life cycle of the virus(Reproduced with permission from New Eng J Med 2004;350:1118-29)
They are transported to the nuclear pores through which the relaxed circular HBV-DNA gains access to the nucleoplasm. There, the relaxed circular HBV-DNA is converted to cccDNA by an unknown mechanism, which however, entails the removal of the covalently attached polymerase, the completion of the (+)-DNA strand and the ligation of the ends of the two DNA strands to form a complete circle. The cccDNA constitutes the transcriptional template for viral mRNA synthesis by the host RNA polymerase II. The newly synthesized transcripts are transported to the cytoplasm where they are translated into the relevant proteins. Following pgRNA encapsidation and synthesis of both DNA strands, the mature nucleocapsids bud through the ER membrane, where the surface proteins are inserted, into the lumen, acquiring in the process their outer envelope. The virions released into the ER lumen are then transported to the hepatocyte cell surface within vesicles, and are released into the circulation. Some of the mature cores may be recycled back to the nucleus and this allows the build up of the cccDNA pool within hepatocytes thus increasing the transcriptional potential of the virus. Virus persistence relies on the cccDNA harboring hepatocytes, the destruction of which is one of the unmet goals in a substantial number of patients undergoing antiviral therapy.
SUBTYPES AND GENOTYPES
The S protein sequence shared by all three envelope proteins as already mentioned contains the a determinant region which constitutes the major immunogenic epitope of the virus. In addition, within this region there are subtypic specificities originally detected by antibodies. The presence of lysine (K) or arginine (R) at position 122 confers d or y specificity respectively. Similarly, specificities w and r are conferred by the presence of K or R at position 160. Moreover, the w subdeterminant can be further divided into w1–w4 specificities. There are nine subtypes of the virus depending on the presence of other subtypic determining amino-acids elsewhere in the a determinant region and the main four are adw, ayw, adr and ayr.
Nucleotide sequencing studies followed by phylogenetic tree analysis have revealed that virus isolates can be separated based on sequence divergence of >8% into eight genotypes designated A–H, with characteristic geographical distribution. More detailed description of these genotypes, including likely differences in relation to pathogenesis and response to antiviral treatment, is given in another chapter of this book.
VARIANTS
Although a DNA virus, HBV replicates through an RNA intermediate which is reverse transcribed, and this step in the replication cycle of the virus is prone to errors by the RNA polymerase II and the viral reverse transcriptase. It is estimated that the HBV genome evolves at a rate of 1.4–3.2 × 10−5 nucleotide substitutions/site/year. As a result of these substitutions, the virus circulates in serum as a population of very closely related genetic variants, referred to as quasispecies. Although a lot of these variants would have mutations that would be deleterious to the virus, as a result of constraints imposed by the overlapping ORFs, some would be advantageous, either offering a replication advantage, or facilitating immune escape (see later chapters).
Finally, genomic analyses have revealed the presence of stable genomic variants that arise during the natural progression of chronic infection. These variants have either reduced levels (core promoter variants) or complete abrogation of HBeAg (precore variants or stop codon variants, G1896A) production. These variants are selected at the time of, or soon after seroconversion to anti-HBe, and become dominant during the reactivation phase (see later chapters). The most common precore mutation is the G1896A substitution, which creates a premature termination codon in the precursor protein from which HBeAg is produced. The double mutation affecting the core promoter region (A1762T, G1764A) is thought to result in decreased transcription of the precore mRNA, with a knock on effect on HBeAg production, whilst pgRNA production remains the same or is even upregulated. It 11is now apparent that additional mutations in this region may contribute to this phenotype.
Finally, in recent years, other variants have been described which arise in response to vaccination. These variants evade the immune response, are not recognized by neutralizing antibodies and may fail to be detected by existing diagnostic tests. In addition, isolates resistant to nucleos(t)ide analogs are responsible for treatment failure in chronic HBV carriers (see later chapters).
CONCLUSION
Since the discovery of the virus, we have learned a lot about its structure, molecular biology and its replication mechanism. There are still however aspects of its biology that remain unknown, and one hopes that in the years to come more speedy progress will be made in this direction. The ultimate goal will be the design of more effective drugs that will reduce significantly, if not eliminate the chronic carrier pool.
FURTHER READING
- Bruss V. Hepatitis B virus morphogenesis. World J Gastroenterol 2007;13:65–73.
- Carman WF, Jazayeri M, Basune A, Thomas HC, Karayiannis P. Hepatitis B surface antigen (HBsAg) variants. In: Thomas HC, Lemon S, Zuckerman AJ (Eds): Viral Hepatitis, 3rd edn, Blackwell Publishing London, 2005;225–41.
- Ganem D, Prince AM. Hepatitis B virus infection—natural history and clinical consequences. N Engl J Med 2004;350:1118–29.
- Karayiannis P, Carman WF, Thomas HC. Molecular variations in the core promoter, precore and core regions of hepatitis B virus, and their clinical significance. In: Thomas HC, Lemon S, Zuckerman AJ (Eds): Viral Hepatitis, 3rd edn, Blackwell Publishing London, 2005;242–62.
- Nassal M. Hepatitis B viruses: reverse transcription a different way. Virus Res 2008;134:235–49.
- Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev 2000;64:51–68.