

SECTION 1
PHYSIOLOGY OF BLOOD
NORMAL HAEMATOPOIESIS
The physiologic process of formation of blood cells is known as haematopoiesis. It proceeds through different stages starting from early embryonic life—mesoblastic stage (yolk sac), hepatic stage, and myeloid (bone marrow) stage. During embryonic and early foetal life, haematopoiesis occurs in the yolk sac (only erythoblasts) and the liver (all blood cells). Blood cell precursors first appear in yolk sac during third week of embryonic development. Definitive haematopoiesis, however, first begins in the mesoderm of intraembryonic aorta/gonad/mesonephros (AGM) region after several weeks. Some blood cell formation also occurs in the spleen (all blood cells), lymph nodes and thymus (mostly lymphocytes). Bone marrow starts producing blood cells around 3rd to 4th month and by birth becomes the exclusive site of blood cell formation (Fig. 1.1). In younger age, whole of the skeletal marrow participates in blood cell production. By late childhood, haematopoiesis becomes restricted to the flat bones such as sternum, ribs, iliac bones and vertebrae and proximal ends of long bones. At other skeletal sites haematopoietic areas are replaced by fat cells. However, when there is increased demand for blood cell production, conversion of yellow fatty inactive marrow to red active marrow can occur. In extremely severe case s (e.g. severe chronic anaemia), resumption of haematopoietic activity in organs other than bone marrow such as liver and spleen (extramedullary haematopoiesis) can occur.


Hierarchy of Haematopoiesis
The scheme of haematopoiesis is shown in Figure 1.2. All blood cells are derived from pleuripotent haematopoietic stem cells, which are present in small numbers in the bone marrow. The haematopoietic stem cell is the most primitive cell in the bone marrow. It has the ability of proliferation, self-renewal, and differentiation along several lineages. The capacity of self-renewal permits life-long continuation of the process. The myeloid and lymphoid stem cells originate from the pleuripotent haematopoietic stem cell. From myeloid and lymphoid stem cells progressively more committed progenitors arise having progressively restricted potential to generate different types of blood cells. Ultimately progenitor cells committed to produce only a single type of cell are derived. These single-lineage progenitors further differentiate to produce morphologically identifiable blood cells (Fig. 1.3). Red cells, granulocytes, monocytes, and platelets are derived from myeloid stem cell while B and T lymphocytes are formed from lymphoid stem cell through intermediate stages.
The factors, which influence the commitment of stem cells and progenitor cells to different lineages, are unknown; the bone marrow microenvironment and responsiveness of progenitors to haematopoietic growth factors appear to play a role.
Haematopoietic Growth Factors (HGFs)
HGFs are a group of proteins that (i) regulate proliferation, differentiation, and maturation of haematopoietic progenitor cells, (ii) influence the commitment of progenitors to specific lineages, and (iii) affect the function and survival of mature blood cells. HGFs are produced by different types of cells, which include T lymphocytes, macrophages, fibroblasts, endothelial cells, and renal interstitial cells (Fig. 1.4 and Table 1.1).
HGFs may bind to specific cell receptors on the surface of the cells to directly induce their proliferation and differentiation or may stimulate the production of other cytokines that then act on the target cells. Two types of HGFs may be distinguished—multilineage HGFs that have action on more than one cell line and lineage-restricted HGFs that act on one specific cell line.

Figure 1.2: Normal haematopoiesis
Abbreviations: SCF: Stem cell factor; TPO: Thrombopoietin; IL: Interleukin; EPO: Erythropoietin; G-CSF: Granulocyte colony stimulating factor; GM-CSF: Granulocyte-macrophage colony stimulating factor; M-CSF: Macrophage colony stimulating factor; CFU-GEMM: Colony-forming unit-Granulocyte Erythroid Megakaryocyte Macrophage; CFU-GM: Colony forming unit-Granulocyte Macrophage; CFU-MegE: Colony forming unit-Megakaryocyte Erythroid; BFU-E: Burst forming unit-Erythroid; CFU-E: Colony forming unit-Erythroid; CFU-G: Colony forming unit-Granulocyte; CFU-M: Colony forming unit-Macrophage; CFU-Eo: Colony forming unit-Eosinophil; CFU-Baso: Colony forming unit-Basophil; NK: Natural killer.


5Examples of multilineage HGFs are GM-CSF (granulocyte macrophage colony stimulating factor) and IL-3 (interleukin-3) while lineage-restricted HGFs are erythropoietin, G-CSF (granulocyte colony stimulating factor), and M-CSF (macrophage colony stimulating factor). For proliferation and differentiation of myeloid progenitors, either GM-GSF or IL-3 and a lineage-specific cytokine (erythropoietin, G-CSF, or M-CSF) are required.
Many of the HGFs have been produced by the recombinant DNA technology and are undergoing clinical trials in various disorders. Recently, recombinant GM-CSF, G-CSF and erythropoietin have been approved for clinical use in certain conditions in USA.
GM-CSF
- GM-CSF stimulates proliferation, differentiation, and maturation of lineages committed to neutrophil and monocyte/macrophage cell lines (CFU-GEMM and CFU-GM) and also enhances the functional activity of mature neutrophils and monocytes.
- Recombinant GM-CSF is used to enhance the myeloid recovery following autologous bone marrow transplantation in non-myeloid malignancies. It is also being used to increase stem cell harvest from peripheral blood in peripheral blood stem cell transplantation. It is being tried in chemotherapy-induced myelosuppression and in myelodysplastic syndrome with neutropaenia.
G-CSF
- G-CSF stimulates myeloid progenitor cells (CFU-G) to form mature neutrophils.
- Recombinant G-CSF is used to reduce duration and severity of neutropaenia in non-myeloid malignancies that are being treated with myelosuppressive chemotherapy and in autologous bone marrow transplantation.
Erythropoietin
- Erythropoietin is a glycoprotein produced in the kidneys (90%) and in the liver (10%). It stimulates progenitor cells committed to erythroid lineage (CFU-E and BFU-E) to proliferate and differentiate.
- It is indicated in patients with anaemia of chronic renal failure who are on dialysis. It is also being tried in zidovudine-treated human immunodeficiency virus-positive patients having anaemia, and in anaemia of cancer.
The Haematopoietic Microenvironment
The existence of haematopoietic microenvironment is suggested by the fact that formation of blood cells is restricted specifically to bone marrow. The exact nature of the microenvironment is poorly understood; however it appears to be composed of endothelial cells, fibroblasts, adipocytes, macrophages, and extracellular matrix.

Figure 1.5: Bone marrow organisation. Bone marrow is located in the intertrabecular space beneath the cortex. Organisation of various cells in the normal bone marrow is characteristic. (1) Endosteal zone: myeloid precursors (myeloblasts, promyelocytes); (2) Intermediate zone: myelocytes, erythroid islands; (3) Central zone: metamyelocytes, bands, segmented neutrophils, erythroid islands, and megakaryocytes
Bone marrow microenvironment provides supporting stroma and growth factors for haematopoiesis. Stem cells and progenitors are bound to the stromal cells or to adhesion molecules within the matrix. Release of mature blood cells from the marrow is regulated by the microenvironment.
Organisation of bone marrow: Organisation of normal bone marrow is shown in Figure 1.5.
RED BLOOD CELLS
Stages of Erythropoiesis
Within the bone marrow, erythroid cells are arranged in the form of islands (Fig. 1.6). The earliest morphologically identifiable erythroid cell in the bone marrow is the proerythroblast (pronormoblast), a large (15–20 μm) cell with a fine, uniform chromatin pattern, one or more nucleoli, and dark blue cytoplasm.
The next cell in the maturation process is the basophilic (early) normoblast. This cell is smaller in size (12–16 μm) and has a coarser nuclear chromatin with barely visible nucleoli. The cytoplasm is deeply basophilic.
The more differentiated erythroid cell is the polychromatic (intermediate) normoblast (size 12–15 μm). The nuclear size is smaller and the chromatin becomes clumped. Polychromasia of cytoplasm results from admixture of blue ribonucleic acid and pink haemoglobin. This is the last erythroid precursor capable of mitotic division.
The orthochromatic (late) normoblast is 8 to 12 μm in size. The nucleus is small, dense and pyknotic and commonly eccentrically-located. The cytoplasm stains mostly pink due to haemoglobinisation. It is called as orthochromatic because cytoplasmic 7staining is largely similar to that of erythrocytes. The nucleus is ultimately expelled from the orthochromatic normoblast with the formation of a reticulocyte. The reticulocyte still has remnants of ribosomal RNA in the form of a cytoplasmic reticulum. After 1 to 2 days in the bone marrow and 1 to 2 days in peripheral blood reticulocytes lose RNA and become mature pink-staining erythrocytes (Figs 1.7 and 1.8).

Figure 1.6: Erythroid island: Within the bone marrow, erythroid progenitors are found in the form of ‘islands’ (erythroid colonies). An erythroid island is composed of erythroblasts surrounding a central macrophage. The more immature precursors are present close to the macrophage and maturing forms are towards the periphery. The macrophage has dendritic processes which extend between erythroid progenitors, support them, and supply iron for haemoglobin synthesis

Figure 1.7: Stages in the formation of a mature red cell. With each stage, cell size and nuclear size become smaller, chromatin clumping increases, and ultimately nucleus is extruded. Colour of cytoplasm gradually changes from basophilic to orange-red

Figure 1.8: Normal peripheral blood smear showing normocytic normochromic—red cells (R), neutrophil (N), eosinophil (E), monocyte (M), small lymphocyte (SL), large lymphocyte (LL), and platelets (P)
About four mitotic divisions and continued differentiation lead to the production of 16 mature erythrocytes from each pronormoblast.
Structure and Function of Erythrocytes
Mature erythrocyte is a round biconcave disc about 7 to 8 μm in diameter. Basic structural properties of various red cell components (haemoglobin, enzymes, and membrane) are outlined below.
Haemoglobin
Haemoglobin is responsible for transport of oxygen from lungs to the tissues and of carbon dioxide from tissues to the lungs. Haemoglobin (MW 64,500 daltons) is composed of haem (consisting of iron and protoporphyrin) and globin. The globin portion of the molecule consists of four (or two pairs of) polypeptide chains. One haem group is bound to each polypeptide chain.
Variants of haemoglobin: Haemoglobin is not homogeneous and normally different variants exist such as A, A2, F, Gower I, Gower II, and Portland (Box 1.1). The last three are present only during embryonic life. Others are present in varying proportions during foetal and adult life. The relative proportions of different haemoglobins are: Adults—HbA 97%, HbA2 2.5%, and HbF 0.5%; Newborns—HbF 80% and HbA 20%.
Haemoglobin A (HbA), the principle haemoglobin of adults, consists of a pair each of alpha (α) and of beta (β) polypeptide chains and its structure is designated as α2β2. Foetal haemoglobin (HbF), the predominant haemoglobin in foetal life, contains a pair of alpha (α) and a pair of gamma (γ) chains. Two types of γ chains are distinguished, Gγ and Aγ, which have different amino acids (either glycine or alanine) at position 136. Thus, HbF is heterogeneous and contains α2γ2 136Gly and α2γ2 136Ala. During embryonic life, there are three haemoglobins: Gower I (ζ2ε2), Gower II (α2ε2) and Portland (ζ2γ2). With foetal development, synthesis of zeta (ζ) and epsilon (ε) chains is replaced by that of α and γ chains, respectively. After birth, production of γ chains switches to that of β and delta (δ) chains.
Structure of globin genes: Normal haemoglobin is a tetramer composed of a pair of α-like and a pair of β-like polypeptide chains. Each chain is linked to one molecule of haem. The α-like polypeptide chains (ζ and α) and β-like polypeptide chains (ε, γ, β, and δ) are encoded by α- and β-globin gene clusters on chromosomes 16 and 11, respectively. The order of genes in α-globin gene cluster from 5’ to 3’ end is ζ-ψζ-ψα2-ψα1-α2-α1. The order of genes in β-globin gene cluster from 5’ to 3’ end is ε-Gγ-Aγ-ψβ-δ-β (Fig. 1.9). The ψζ, ψα2, ψα1, and ψβ are pseudogenes.

Figure 1.9: α and β globin gene clusters. Open boxes represent pseudogenes while filled boxes represent active genes. Normal genotype is shown below each gene cluster

A pseudogene (ψ) contains sequences similar to a functional gene but is rendered inactive due to mutation during evolutionary process.
In humans, autosomal chromosomes occur in pairs. As each member of chromosome 16 has two α gene loci (a locus refers to specific physical position of a gene on chromosome), there are total four α genes. However, there is only one β globin gene locus on chromosome 11, and therefore β genes are two in number.
Genes are the base sequences, which are present along the DNA strands and are necessary for the formation of a protein. The different functional areas of a globin gene are:
- Exons and introns: The regions of DNA strand which encode amino acids in the protein product are known as exons while non-coding regions which interrupt the coding sequences are known as introns or intervening sequences. Each globin gene contains three exons and two introns.
- Splice junction sequences: These are sequences at the junction of exons and introns and are required for precise splicing (or removal) of introns during the formation of mRNA.
- Promoter: The promoter region is present towards 5’ end of the gene and contains sequences to which the RNA polymerase binds; it is necessary for correct initiation of transcription. Two promoter sequences are TATA and CCAAT.
- Polyadenylation signal: The 3’ end of the globin gene contains the sequence AATAAA that serves as a signal for the addition of a poly-A track to the mRNA transcript (Fig. 1.10).
Steps in the synthesis of globin: Globin synthesis involves three steps—transcription, processing of mRNA, and translation (Fig. 1.11).
- Transcription: Transcription involves synthesis of a single strand of RNA from DNA template by the enzyme RNA polymerase. The base sequence of RNA, which is produced, is complementary to the base sequence of DNA. Binding of RNA polymerase to the promoter is essential for accurate initiation of transcription. RNA polymerase slides along the DNA strand in a 5’ to 3’ direction and builds the RNA molecule. Transcription continues through exons and introns and when a chain terminating sequence is encountered, RNA polymerase gets separated from the DNA strand. The RNA strand thus formed is called as messenger RNA (mRNA).
- Processing of mRNA: In the next stage, mRNA molecule is processed by addition of a cap structure and a poly-A tail and by removal of introns. A cap structure (modified nucleotides) is added at the 5’ end of mRNA; though the exact role is unknown, capping appears to be necessary for initiation of translation. At the 3’ end a poly-A tail consisting of about 150 adenylic acid residues is added. AAUAAA sequence at the 3’ end signals the addition of poly-A tail about 20 bases downstream from the polyadenylation site. Polyadenylation is required for stability of the transcript and its transport to the cytoplasm. Excision of introns and joining together of exons in the mRNA transcript are essential before mRNA is transported from the nucleus to the cytoplasm.Accurate splicing is guided by the presence of GT dinucleotide at the exon-intron boundary (5’ end of intron) and AG dinucleotide at the intron-exon boundary (3’ end of intron). Intron 1 is excised before intron 2. During splicing, excision at 5’ exon-intron boundary occurs initially with the formation of ‘lariat’ structures; subsequently excision at the 3’ intron-exon boundary occurs followed by joining of exons.
- Translation: This process, which occurs on ribosomes, consists of synthesis of a polypeptide chain according to the directions provided by the mRNA template. There are three kinds of RNA which take part in the synthesis of polypeptides—messenger RNA (mRNA), transfer RNA (tRNA) and ribosomal RNA (rRNA). The mRNA, transcribed from the DNA template, carries the genetic code from the nucleus to the cytoplasm and determines the sequence of amino acids in the formation of a polypeptide. The tRNA transports specific amino acids from the cytoplasm to the specific locations (codons) along the mRNA strand; each tRNA binds and transports a specific amino acid. The rRNA, along with certain structural proteins, constitutes the ribosome which serves as a site for protein synthesis.

The different steps of protein synthesis (translation) are activation, initiation, elongation, and termination. In activation, an amino acid combines with its specific tRNA molecule in the cytoplasm; such tRNA is called as activated or charged tRNA. Translation always begins at a codon that specifies methionine (AUG, the initiator codon). The process of translation is initiated when a methionine-bearing specific tRNA binds with initiator codon in mRNA. Elongation of polypeptide chain occurs when successive amino acids are added after methionine according to the pattern provided by the genetic code. During this process, movement of ribosomes occurs along the mRNA strand and ribosome slides to the next codon when an amino acid specified by preceding codon is added to the growing polypeptide chain. Amino acids are attached to each other by peptide bonds. Termination of translation occurs when a chain-terminating (or a stop) codon is encountered (UAA, UAG, or UGA). This is followed by release of the completed polypeptide chain from the ribosomes.
The primary polypeptide chain is then organised into a secondary and a tertiary structure from interactions in its amino acids. One molecule of haem is attached to each polypeptide chain. Two different pairs of polypeptide chains with their attached haem moieties associate with each other to form a tetrameric haemoglobin molecule.
Changes in globin gene expression during development (Globin ‘switching’): Hb Gower I, Hb Gower II, and Hb Portland are the predominant haemoglobins during embryonic life (upto 12 weeks). HbF (α2γ2) is the major haemoglobin of foetal life; it starts gradually declining after 36 weeks of gestation and constitutes less than 1% of haemoglobin in adults. Beta (β) chain synthesis starts around 10th week of gestation and is significantly augmented around the time of birth. HbA (α2β2) gradually becomes the predominant haemoglobin by 3 to 4 months of age. Delta (δ) globin gene is expressed late in the third trimester but HbA2 (α2δ2) remains at a low level (about 2.5%) in adults.
The developmental changes in the expression of the globin genes can be correlated with the time of appearance of clinical features in haemoglobinopathies. Thus, α-thalassaemia manifests at birth while clinical features of β-thalassaemia appear a few months after birth.
Biosynthesis of haem: Haem is a complex of protoporphyrin and iron. Biosynthesis of haem requires mitochondrial (as well as cytosolic) enzymes and therefore only erythroid precursors but not mature red cells can synthesize haem.
Structure and function of haemoglobin: Haemoglobin is a tetramer composed of four polypeptide chains (α1, α2, β1, and β2) and four haem groups. α chain consists of 141 amino acids while β chain has 146 amino acids. Each polypeptide chain is arranged in a helical conformation. There are eight helical segments designated A to H. Iron of haem is covalently bound to histidine at the eighth position of the F helical segment. Charged or polar residues are arranged on the outer surface while the uncharged or nonpolar residues are arranged towards the inner part of the molecule. Haem is suspended in a ‘pocket’ formed by the folding of the polypeptide chain and residues in contact with haem are nonpolar. The four polypeptide chains make contact at α1β1 and α1β2 interfaces. The former is a stabilizing contact while the latter is the functional contact across which movement of chains occurs during oxygenation and deoxygenation.
The function of haemoglobin is transport of oxygen from the lungs to the tissues. As partial pressure of oxygen increases, haemoglobin shows progressively increasing affinity for oxygen. When first oxygen binds to the haem group, it successively increases the oxygen affinity of the remaining three haem groups. When the percent saturation of haemoglobin with oxygen is plotted against the partial pressure of oxygen, a sigmoid-shaped oxygen dissociation curve is obtained. Small changes in oxygen tension allow significant amount of oxygen to be released or bound.
Factors affecting oxygen affinity of haemoglobin are pH, temperature, intraerythrocyte level of 2,3-diphosphoglycerate (2,3-DPG) and presence of haemoglobin variants. The Bohr effect refers to the alteration in oxygen affinity due to alteration in pH. Low pH (e.g. in tissues) reduces the oxygen affinity while higher pH (e.g. in lungs) increases the oxygen affinity of haemoglobin. High temperature reduces the oxygen affinity while low temperature increases the oxygen affinity. 2,3-DPG binds to deoxyhaemoglobin with considerably more affinity than to oxyhaemoglobin and stabilizes the deoxyhaemoglobin state. Low levels of 2,3-DPG in red cells in stored blood in blood bank are associated with reduced release of oxygen after blood transfusion. Haemoglobin variants with high oxygen affinity are methaemoglobin, Hb Bart's, and Hb H.
Red Cell Enzymes
The mature red cell requires energy to preserve the integrity of the cell membrane, for active transport of cations, for nucleotide salvage, and for synthesis of glutathione. This is mostly provided by glycolysis (Embden-Meyerhof pathway). In this metabolic pathway, glucose is converted to pyruvate and lactate through a series of enzymatic reactions 13with generation of ATP (Fig. 1.12). In the middle of the glycolytic pathway, a Rapoport-Luebering shunt exists in red cells for the synthesis of 2,3-DPG. The net yield of ATP from glycolysis is dependent upon the amount of glucose utilised by this shunt. 2,3-DPG is an important determinant of the oxygen affinity of haemoglobin. Apart from ATP and 2,3-DPG, another important product of glycolysis is NADH that is required for reduction of methaemoglobin to oxyhaemoglobin.
The aerobic hexose monophosphate shunt (pentose phosphate shunt) is another metabolic pathway in red cells. The two dehydrogenase enzymes, glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (6-PGD), cyclically generate NADPH from NADP.

14These two enzymes also convert glucose-6-phosphate to pentose, which is returned to the main glycolytic pathway. NADPH and the enzyme glutathione reductase are required for the regeneration of reduced glutathione (GSH) from oxidised glutathione (GSSG). GSH along with glutathione peroxidase detoxifies hydrogen peroxide and protects haemoglobin from oxidant damage.
Most of the methaemoglobin produced in the normal cell is reduced to haemoglobin by NAD-linked methaemoglobin reductase. Methaemoglobin reductase that is linked to NADP requires methylene blue for its activation and is more effective in drug-induced methaemoglobinaemia (Fig. 1.12).
Various metabolic pathways in the red cell are summarised in Box 1.2.
Red Cell Membrane
The red cell membrane (Fig. 1.13) is composed of lipids, a complex network of proteins, and a small amount of carbohydrates. The membrane lipids include phospholipids, cholesterol, and glycolipids. The phospholipids are arranged in the form of a bilayer. The distribution of phospholipids is asymmetrical with aminophospholipids and phosphatidyl inositols located preferentially in the inner part of the bilayer and choline phospholipids in the outer part. The polar head groups are oriented both internally and externally while the fatty acid chains are oriented toward each other. The red cell membrane proteins are embedded within the lipid bilayer (transmembranous proteins) and also form an extensive network beneath the bilayer (submembranous proteins). The transmembranous and submembranous proteins constitute the red cell cytoskeleton. Red cell membrane proteins can be separated according to molecular size by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE). Different bands can be visualised when stained with a protein stain such as Coomassie blue. The important skeletal proteins are spectrin (bands 1 and 2), ankyrin (band 2.1), anion exchange protein (band 3), protein 4.1, and actin (band 5). Spectrin is the major cytoskeletal protein; it consists of two dissimilar chains, alpha and beta, which are intertwined together. The head ends of the spectrin dimers interact with those of the other spectrin dimers to form spectrin tetramers and oligomers. The tail ends of spectrin tetramers interact with actin and this association is stabilised by protein 4.1. On electron microscopy, the skeletal proteins appear to be organised in the form of a hexagonal lattice; the arms of the hexagon are formed by spectrin and corners by actin, protein 4.1, and adducin.

The anchorage of the cytoskeleton to the overlying lipid bilayer is achieved by two associations: band 3-ankyrin-spectrin association and glycophorin C-protein 4.1 association. Band 3 is the anion exchange channel through which the exchange of HCO3− and Cl¯ occurs.
The membrane provides mechanical strength and flexibility to the red cell to withstand the shearing forces in circulation. The cell membrane also serves to maintain the red cell volume by the cation pump. The cation pump, operated by the membrane enzyme ATPase, regulates the intracellular concentration of Na+ and K+. The membrane ATPase also drives the calcium pump, which keeps the intracellular Ca++ at a very low level. The red cells exchange HCO3¯ (formed from tissue CO2) in the lungs with Cl¯ through the anion exchange channel (band 3) in the membrane.
Red cell destruction: The life span of normal erythrocytes is about 120 days. The senile red cells are recognized by macrophages of reticuloendothelial system and are destroyed mainly in the spleen. Globin is converted to amino acids, which are stored to be recycled again. Degradation of haem liberates iron and porphyrin. Iron is stored as ferritin in macrophages or is released in circulation where it is taken up by transferrin and transported to erythroid precursors in bone marrow. The porphyrin is converted to bilirubin.
WHITE BLOOD CELLS
Neutrophils
Stages of Granulopoiesis
The maturation sequence in granulopoiesis is—myeloblast, promyelocyte, myelocyte, metamyelocyte, band cell, and segmented granulocyte (Fig. 1.14). This process occurs within the marrow.
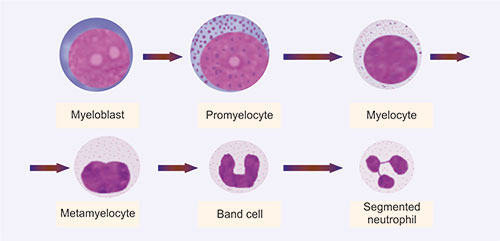
Myeloblast: Myeloblast is the earliest recognizable cell in the granulocytic maturation process. It is about 15 to 20 μm in diameter, with a large round to oval nucleus, and small amount of basophilic cytoplasm. The nucleus contains 2 to 5 nucleoli and nuclear chromatin is fine and reticular.
Promyelocyte: The next stage in the maturation is promyelocyte which is slightly larger in size than myeloblast. Primary or azurophil granules appear at the promyelocyte stage. The nucleus contains nucleoli as in myeloblast stage, but nuclear chromatin shows slight condensation.
Myelocyte: Myelocyte stage is characterised by the appearance of secondary or specific granules (neutrophilic, eosinophilic, or basophilic). Myelocyte is a smaller cell with round to oval eccentrically placed nucleus, more condensation of chromatin than in promyelocyte stage, and absence of nucleoli. Cytoplasm is relatively greater in amount than in promyelocyte stage and contains both primary and secondary granules. Myelocyte is the last cell capable of mitotic division.
Metamyelocyte: In the metamyelocyte stage, the nucleus becomes indented and kidneyshaped, and the nuclear chromatin becomes moderately coarse. Cytoplasm contains both primary and secondary granules.
Band stage (stab form): This is characterised by band-like shape of the nucleus with constant diameter throughout and condensed nuclear chromatin.
Segmented neutrophil (polymorphonuclear neutrophil): With Leishman's stain, nucleus appears deep purple with 2 to 5 lobes which are joined by thin filamentous strands. Nuclear chromatin pattern is coarse. The cytoplasm stains light pink and has small, specific granules (Fig. 1.15).
Primary and secondary granules: The neutrophil granules are of two types: primary or azurophilic granules and secondary or specific granules. Azurophil granules contain myeloperoxidase, lysozyme, acid phosphatase, elastases, collagenases, and acid hydrolases.


Figure 1.16: Neutrophil kinetics. After release from the marrow, neutrophils in peripheral blood can be divided into two compartments: circulating pool (measured by leucocyte count) and marginating pool (neutrophils adhering to endothelium via adhesion molecules; this portion is not measured by leucocyte count)
Specific granules contain lysozyme, lactoferrin, alkaline phosphatases, vitamin B12-binding protein and other substances.
Function of neutrophils: After their formation, neutrophils remain in marrow for 5 more days as a reserve pool. Neutrophils have a life span of only 1 to 2 days in circulation. Neutrophil compartments and kinetics is shown in figure 1.16.
In response to infection and inflammation, neutrophils come to lie closer to endothelium (margination) and adhere to endothelial surface (sticking). This is followed by escape of neutrophils from blood vessels to extravascular tissue (emigration). The escape of neutrophils is guided by chemotactic factors present in the inflammatory zone. Chemotactic factors for neutrophils include bacterial factors, complement components such as C3a and C5a, breakdown products of neutrophils, fibrin fragments, and leukotriene B4. Phagocytosis follows which involves three steps—antigen recognition, engulfment, and killing of organism. Neutrophils have receptors for Fc portion of immunoglobulins and for complement. Many organisms are identified by neutrophils after they are coated with opsonins (IgG1, IgG3, and C3b). Cytoplasm of the neutrophil extends in the form of pseudopods around the microorganism, and the organism is eventually completely enclosed within the membrane-bound vacuole (phagosome). Lysosomal 18granules fuse with phagosome and discharge their contents into the phagolysosome. The last step in phagocytosis is killing of micro-organism, which may be either oxygen-dependent or oxygen-independent. Oxygen-dependent mechanism involves conversion of oxygen to hydrogen peroxide by oxidase in phagolysosome; myeloperoxidase in the presence of halide ion (e.g. Cl¯) converts hydrogen peroxide to HOCl· that has a strong bactericidal activity.
Another oxygen-dependent bactericidal mechanism is independent of myeloperoxidase and involves formation of superoxide radicals. Oxygen independent bactericidal mechanism occurs in lysosomal granules and is mediated by substances such as lysozyme, major basic protein, bactericidal permeability increasing protein, etc.
Eosinophils
Eosinophil forms via same stages as the neutrophil and the specific granules first become evident at the myelocyte stage. The size of the eosinophil is slightly greater than that of neutrophil. The nucleus is often bilobed and the cytoplasm contains numerous, large, bright orange-red granules. The granules contain major basic protein, cationic protein, and peroxidase (which is distinct from myeloperoxidase). Eosinophilic peroxidase along with iodide and hydrogen peroxide may be responsible for some defense against helminthic parasites. Crystalloids derived from eosinophil membrane form characteristic Charcot-Leyden crystals. Maturation time for eosinophils in bone marrow is 2 to 6 days and half-life in blood is less than 8 hours. In tissues, they reside in skin, lungs, and GIT.
Basophils
Basophils are small, round to oval cells which contain very large, coarse, deep purple granules. The nucleus has condensed chromatin and is covered by granules.
Mast cells in connective tissue or bone marrow differ morphologically from basophils in following respects: mast cells (10–15 μm) are larger than basophils (5–7 μm); mast cells have a single round to oval eccentrically placed nucleus while nucleus of the basophils is multilobed; and the cytoplasmic granules in mast cells are more uniform. Tissue mast cells are of mesenchymal origin.
Basophil granules contain histamine, chondroitin sulfate, heparin, proteases, and peroxidase. Basophils bear surface membrane receptors for IgE. Upon reaction of antigen with membrane-bound IgE, histamine and other granular contents are released which play a role in immediate hypersensitivity reaction. Basophils are also involved in some cutaneous basophil hypersensitivity reactions.
Monocytes
The initial cell in development is monoblast, which is indistinguishable from myeloblast. The next cell is promonocyte which has an oval or clefted nucleus with fine chromatin pattern and 2 to 5 nucleoli. The monocyte is a large cell (15–20 μm), with irregular shape, oval or clefted (often kidney-shaped) nucleus and fine, delicate chromatin.

Cytoplasm is abundant, blue-grey with ground glass appearance and often contains fine azurophil granules and vacuoles.
Monocytes circulate in blood for about 1 day and then enter and settle in tissues where they are called as macrophages or histiocytes. In some organs, macrophages have distinctive morphologic and functional characteristics (Fig. 1.17).
Macrophage phagocytosis is slower as compared to neutrophils. Macrophages have receptors for Fc portion of IgG and C3b and cause phagocytosis of organisms that are coated with these substances. Macrophages also recognise and phagocytose some target substances by their surface characteristics.
Macrophages may be activated by certain stimuli such as lymphokines (interferon γ secreted by T lymphocytes), direct contact with micro-organism, phagocytized material and complement components. Activated macrophages are larger and have enhanced metabolic and phagocytic activity.
Activated macrophages secrete a variety of biologically active substances:
- Cytokines—interleukin-1, tumour necrosis factor α, interferons α and β;
- Growth factors—fibroblast growth factors, haematopoietic growth factors (GM-CSF and G-CSF), angiogenesis factor, transforming growth factor β;
- Complement proteins;
- Coagulation factors, e.g. thromboplastin;
- Oxygen-derived free radicals—hydrogen peroxide, superoxide, hydroxyl radical;
- Prostaglandins and leukotrienes which are chemical mediators in inflammation;
- Enzymes—elastases, collagenases, lysozyme, plasminogen activator, lipases;
- Fibronectin;
- Transferrin, transcobalamin II, apolipoprotein E.
The major functions of macrophages are processing and presentation of antigens to T lymphocytes during immune response, killing of intracellular pathogens, tumoricidal activity, and phagocytosis of organisms and of injured and senescent cells.
Lymphocytes
These are of two types—small and large. Most of the lymphocytes in peripheral blood are small (7–10 μm). The nucleus is round or slightly clefted with coarse chromatin and occupies most of the cell. The cytoplasm is basophilic, slight and is visible as a thin border around the nucleus.
Around 10 to 15% of lymphocytes in peripheral blood are large (10–15 μm). Their nucleus is similar to that of small lymphocytes but their cytoplasm is relatively more and contains few azurophilic (dark red) granules.
On immunophenotyping, there are two major types of lymphocytes in peripheral blood: B lymphocytes (10–20%) and T lymphocytes (60–70%). Differences between B and T lymphocytes are presented in Table 1.2. About 10 to 15% of lymphocytes are of natural killer (NK) cell type.
B Lymphocytes
B lymphocytes arise from the lymphoid stem cells in the bone marrow. Initial development occurs in primary lymphoid organ (bone marrow) from where B cells migrate to the secondary lymphoid organs (lymph nodes and spleen) where further differentiation occurs on antigenic stimulation. On activation by antigen, B cells undergo differentiation and proliferation to form plasma cells and memory cells. Plasma cells secrete immunoglobulins while memory cells have a lifespan of many years and upon restimulation with the same antigen undergo proliferation and differentiation. Plasma cell is a round to oval cell with eccentrically placed nucleus and deeply basophilic cytoplasm. Nuclear chromatin is dense and arranged in a radiating or cartwheel pattern. The function of B lymphocytes is production of antibodies after differentiation to plasma cells. Antibodies can cause destruction of target cells/organisms either directly or by opsonisation.
|
Immunoglobulin gene rearrangement: There are 5 classes of immunoglobulins: IgM, IgG, IgA, IgD, and IgE. Each immunoglobulin molecule consists of two heavy chains and two light chains. Heavy chain (μ, δ, γ, ε, and α) determines the class of the immunoglobulin molecule. The two light chains are kappa (κ) and lambda (λ). Both heavy and light chains have constant and variable regions. The antigen-specificity of a particular immunoglobulin molecule depends upon amino acid sequence in the variable region (antigen-binding site). To react with a vast array of antigens, the immune system must have the capability to produce a large number of antigen-specific variable regions. The amino acid sequences in constant regions of heavy and light chains remain same for particular class and do not determine antigen specificity.
The heavy chain genes are located on chromosome 14. Light chain genes are located on chromosomes 2 (κ chain) and 22 (λ chain).
An immunoglobulin gene consists of V (Variable) and J (Joining) exons which code for amino acid sequences in variable region, and C (Constant) exon which codes for amino acid sequences in constant region. In heavy chain genes, another exon called D (Diversity) is present which codes for amino acids in variable region (in addition to V and J) (Fig. 1.18). There are several gene segments in V, D, and J regions, and therefore numerous antigen specificities can arise by various combinatorial rearrangements.
The unrearranged heavy and light chain genes are present in all the cells of the body (germ-line configuration); complete rearrangement occurs only in B cells. During development of B cells, rearrangement of heavy chain genes precedes the rearrangement of light chain genes.
Heavy chain gene rearrangement (Fig. 1.18): First, a D gene segment combines with a J segment (to form DJ), followed by combination of DJ with a V gene segment. The VDJ thus formed codes for amino acid sequence in variable region. From the C region, initially Cμ segment (which is located immediately 3’ to the VDJ exon) is transcribed so as to form VDJCμ mRNA. This causes expression of μ heavy chains in the cytoplasm of pre-B cells, and after rearrangement of light chain genes expression of IgM on the surface of early B cells. Usually Cδ locus, which lies very close to Cμ locus, is also transcribed so that the cell expresses both IgM and IgD (with identical variable region sequences) on the surface.
Light chain gene rearrangement: During light chain gene rearrangement, initially a VJ exon is formed by fusion of one V and one J segment. The VJ exon is transcribed along with C exon and after splicing forms VJC mRNA.
Second immunoglobulin gene rearrangement can occur in activated B cells in which switching to new C segment of heavy chain gene occurs, i.e. Cμ to Cγ1 or Cα1 or Cε, etc. This causes change in the class of the immunoglobulin molecule, i.e. IgM to IgG1 or IgA or IgE, etc. Switching does not affect VDJ exon so that antigen specificity is not altered.
B cell ontogeny (Fig. 1.19): During B cell development, sequential genotypic and phenotypic changes occur which can be detected by immunological markers and gene rearrangement studies. Important features in B cell ontogeny are outlined below.
- There are two stages of B cell development: antigen-independent and antigen-dependent. Antigen-independent development occurs in bone marrow while antigen-dependent development occurs in peripheral lymphoid tissues.Figure 1.18: Immunoglobulin heavy chain gene rearrangement. DJ rearrangement occurs first followed by VDJ joining (V3D2J3 in this example). V and D regions other than V3 and D2 are deleted. The rearranged gene is transcribed into μRNA and intervening sequences between J3 and Cμ are spliced. The mRNA formed is translated into a μ heavy chain in cytoplasm
- Rearrangement of immunoglobulin genes and immunoglobulin expression: Initially there is rearrangement of heavy chain genes which is followed by rearrangement of light chain genes. In pre-B cell, rearrangement of heavy chain gene causes appearance of μ heavy chain in cytoplasm (Cμ). This is followed by rearrangement of light chain genes. Light chains associate with μ heavy chain in cytoplasm and IgM is expressed on the cell surface. Mature B cells express both IgM and IgD. In activated B cells, class switching of heavy chains occurs such as IgM to IgG or IgA or IgE. Plasma cells do not have surface expression of immunoglobulin but synthesize and secrete large amounts of immunoglobulins of one class.
- Cell surface antigens: The earliest antigens expressed during B cell development are TdT (within the nucleus) and HLA-DR (on cell surface); these are, however, not specific for B cells. There is a sequential appearance of antigens on developing B cells: CD19, CD10, and CD20. With development and maturation new antigens are expressed while some of the previous ones are lost. Plasma cells express specific antigens such as CD38 (Fig. 1.19).
- According to the fundamental theory of lymphoid neoplasms, the neoplastic cells represent cells arrested at various stages of normal lymphocyte development.

T lymphocytes
T lymphocytes originate from the progenitor cells in the bone marrow and undergo maturation in thymus. After their release from thymus, T cells circulate in peripheral blood and are transported to secondary lymphoid organs (i.e. paracortex of lymph nodes and periarteriolar lymphoid sheaths in spleen).

Figure 1.19: Normal stages of B cell development showing sequential expression of various antigens and heavy and light chain gene rearrangement. As shown at the bottom, lymphoid neoplasms represent cells arrested at various stages of normal development
Abbreviations: CLL/SL: Chronic lymphocytic leukaemia/small lymphocytic lymphoma; DLBCL: Diffuse large B cell lymphoma; MALT: Mucosa associated lymphoid tissue.
There are two major subsets of mature T cells: T helper-inducer cells and T cytotoxic cells. Helper-inducer T cells regulate the functions of B cells and cytotoxic T cells. T helper-inducer cells recognise antigen presented by antigen-presenting cells in association with MHC class II molecules. Cytotoxic T cells recognize antigen in association with MHC class I molecules and play an important role in cell-mediated immunity. T lymphocytes secrete cytokines such as interferon-γ, GM-CSF, tumour necrosis factor, and certain interleukins.
T cell receptor (TCR): The T cell receptor complex consists of seven polypeptide chains. In the majority (95%) of T cells, α and β chains form the antigen-binding site of TCR (αβ TCR); each of these chains has a variable and a constant region similar to immunoglobulins. α and β chains are linked together by a disulfide bond to form α-β heterodimer. The α-β heterodimer is non-covalently associated with CD3 molecular complex which is composed of five polypeptide chains (Fig. 1.20). The variable regions of α and β chains bind antigen, while CD3 converts this antigenic recognition into intracellular activating signals.

In a minority of T cells, γ and δ polypeptide chains are present instead of α and β chains (γδ TCR).
TCR gene rearrangement: The genetic structure of TCR bears resemblance to that of immunoglobulins. The TCR β chain gene is located on chromosome 7 and TCR α chain gene is located on chromosome 14.
Although all somatic cells contain T cell receptor gene in germ-line configuration, rearrangement occurs only in T cells. The TCR β gene consists of variable (V), diversity (D), joining (J), and constant (C) regions. One segment each from V, D, and J regions join together with deletion of intervening sequences. The rearranged gene is transcribed into mRNA. Splicing in transcribed mRNA causes fusion of VDJ to C region to generate TCR β mRNA. Rearrangement of other polypeptide chain occurs similarly. As there are a number of V, D, and J segments which code for amino acid sequences in variable region, it is possible to generate T cell receptors with different antigen specificities by various combinations during rearrangement.
Rearrangement of TCR β gene precedes the rearrangement of TCR α gene.
T cell ontogeny (Fig. 1.21): Progenitor T cells from the bone marrow are transported to thymus where they undergo maturation. During maturation, there is rearrangement of TCR genes, expression of some cell surface proteins, and acquisition of ability to distinguish self-antigen from foreign antigens.
Initially, immature cortical thymocytes express CD7, TdT, and cytoplasmic CD3 (cCD3). Those T cells which subsequently are going to form α and β polypeptides (αβ TCR) first rearrange TCR β gene followed by TCR α gene. Expression of αβ TCR occurs in association with expression of CD3 on surface of cells. Initially, both CD4 and CD8 antigens are acquired; with further maturation cell retains either CD4 or CD8 antigen. CD4+ cells are called as helper-inducer T cells whereas CD8+ cells are called as cytotoxic T cells. The mature T cells are released from thymus, circulate in peripheral blood, and are transported to peripheral lymphoid organs.

Figure 1.21: Stages of T cell development. Correlation of stages with T cell neoplasms is shown at the bottom
Natural Killer (NK) Cells
About 10 to 15% of peripheral blood lymphocytes are natural killer cells. These cells do not require previous exposure or sensitisation for their cytotoxic action. They play a significant role in host defense against tumour cells and virally-infected cells. Morphologically, these cells are large granular lymphocytes.
White Cell Antigens
The HLA System
The HLA or human leucocyte antigens are encoded by a cluster of genes on short arm of chromosome 6 called as major histocompatibility complex (MHC). There are numerous allelic genes at each locus which makes the HLA system extremely polymorphic. The antigens are called as HLA because they were first detected on white blood cells, although they are present on several other cells also.
Types of HLA antigens: There are three types of HLA antigens: class I, class II, and class III.
Class I antigens: Genes at HLA-A, HLA-B, and HLA-C positions specify class I antigens. Class I antigens are glycoproteins which are associated noncovalently with β2 microglobulin. Almost all nucleated cells possess class I antigens (Fig. 1.22).
Class II antigens: HLA-D region (HLA-DR, HLA-DQ, and HLA-DP) encodes class II antigens. These consist of two glycoprotein chains α and β which are bound noncovalently. Class II antigens are present on monocytes, macrophages, B-lymphocytes, and stimulated T lymphocytes (Fig. 1.22).

Class III antigens: Genes specifying class III antigens are situated between genes which specify class I and class II antigens. Class III genes encode certain complement components and cytokines (tumour necrosis factor).
The HLA genes are closely linked and are inherited by an individual as a haplotype from each parent. In a given population, certain HLA haplotypes occur much more frequently than expected by chance alone (linkage disequilibrium).
Significance of HLA antigens: (1) They are important as histocompatibility antigens in organ transplantation; (2) HLA antigens play a major role in recognition of foreign antigens and in immunity; (3) In transfusion medicine, HLA antigens are responsible for alloimmunization against platelet antigens and refractoriness to platelet transfusions, febrile transfusion reactions, and graft-verus-host disease; (4) A relationship exists between presence of some HLA antigens and susceptibility to certain diseases; (5) HLA antigen typing can be used for paternity testing.
Tests for detection of HLA antigens:
- Lymphocytotoxicity test: Class I HLA antigens are detected by lymphocytotoxicity test. In this test, lymphocytes are first isolated from peripheral blood by density gradient separation. These lymphocytes are then added to known specific antisera in microwell plates and incubated to allow the antibodies to bind to target antigens. Complement is added to the lymphocyte-antiserum mixture followed by further incubation. If particular antigen is present on lymphocytes, then antigen-antibody reaction occurs which activates and fixes the complement, leading to cell membrane injury and cell death. A vital dye (eosin Y or trypan blue) is then added to differentiate living 27from dead cells. Damaged cells take up the dye due to the increased permeability of injured cell membrane while living cells remain unstained.For detection of class II antigens (HLA-DR and HLA-DQ), lymphocytotoxicity test is carried out on B lymphocytes. This is because class II antigens are present on B lymphocytes and not on unstimulated T cells. Separation of B lymphocytes is usually achieved by magnetic beads, which are coated with monoclonal antibodies against B cells.
- Mixed lymphocyte culture (MLC) or mixed lymphocyte reaction (MLR): This test is used for detection of class II antigens. Lymphocytes from two different individuals are cultured together. Lymphocytes from one individual are inactivated by irradiation or by mitomycin C before the test to suppress their division; these lymphocytes are called stimulator cells. During incubation in culture, lymphocytes from the other individual recognise the foreign class II HLA antigens on stimulator cells and respond by enlarging in size, synthesizing DNA, and proliferating (blastogenic response); these cells are called as responder cells. If HLA class II antigens on responder and stimulator cells are identical, there is no blastogenic response. After 5 to 7 days 3H-thymidine is added to the culture and radioactive material incorporated into the dividing (responder) cells is quantitated. The amount of radioactive thymidine incorporated into the dividing cells is proportional to DNA synthesis.
- Primed lymphocyte typing (PLT) test: This test is used for detection of HLA-DP antigens. It is based on mixed lymphocyte culture. In this method the culture of lymphocytes is extended for 2 weeks during which death of stimulator cells occurs and proliferation of responder cells halts. As these responder cells have been primed (i.e. sensitised), their re-encounter with the cells, which carry the same HLA-DP antigen as the initial stimulator cells, causes their rapid proliferation.
- DNA analysis: Allelic genes at HLA-D loci can be identified by allele-specific oligonucleotide probe analysis. With the advent of polymerase chain reaction technology, which amplifies the desired DNA sequence several times, sensitivity of DNA analysis for HLA typing has greatly increased.
Neutrophil-specific Antigens
Apart from HLA antigens, granulocytes also possess neutrophil-specific antigens. These are NA1, NA2, NB1, NB2, NC1, ND1, NE1, 9a, and HGA-3a, 3b, 3c, 3d and 3e. Neutrophil-specific antigens play an important role in immune neutropaenias and in febrile nonhaemolytic transfusion reactions.
IMMUNE SYSTEM
As white cells play a major role in immunity, it is appropriate to consider antibodies and complement here.
Antibodies
Antibodies are immunoglobulins that react with antigens. They are produced by plasma cells, which in turn are derived from B lymphocytes.
Structure of Immunoglobulins
The immunoglobulin molecule consists of two identical heavy (H) chains and two identical light (L) chains. The H and L chains are linked together by disulfide (s-s) bonds. Five classes of immunoglobulins are recognised based on the type of H chain: IgA (α or alpha H chain), IgD (δ or delta), IgE (ε or epsilon) IgG (γ or gamma), and IgM (μ or mu). Light chains are of two varieties—κ (kappa) and λ (lambda).
A molecule of immunoglobulin consists of light chains of the same type (either κ or λ); both types of light chains are never present together. Kappa and lambda chains are present in 2:1 proportion in immunoglobulins.
Each chain has a constant and a variable region (Fig. 1.23). Amino acid composition in the carboxy terminal region of heavy chain and light chain is the constant region; in the heavy chain it determines the class of the immunoglobulin molecule. The CH2 domain in IgG binds complement while CH3 domain binds to Fc receptor of monocytes. The variable region of the molecule (VL and VH) is the specific antigen-binding site and is in the amino-terminal part of the molecule. The area J of the heavy chains in the constant regions between CH1 and CH2 domains is flexible and is called hinge region; due to this the two antigen-binding sites can move in relation to each other spanning variable distances.
Each immunoglobulin molecule can be digested by a proteolytic enzyme papain just above the disulphide bond joining the two heavy chains into three parts: one Fc and two Fab fragments. The fragment, which contains the carboxy terminal and constant parts of both heavy chains, is called the Fc (fragment crystallizable) fragment. Each Fab (fragment antigen binding) fragment contains amino terminal portion of H chain and complete light chain and has the antigen-combining site (Fig. 1.23).

Classes of Immunoglobulins
IgG: This is the major immunoglobulin in plasma comprising about 75% of all circulating immunoglobulins. IgG is the monomer of the basic immunoglobulin structure. There are four subclasses of IgG: IgG1, IgG2, IgG3, and IgG4. Relative concentration in serum can be represented as IgG1 >lgG2>lgG3>lgG4. IgG is usually produced during secondary immune response. It is the only immunoglobulin, which is transferred transplacentally to the foetus from the mother. The foetus cannot synthesize IgG and therefore IgG antibodies in the newborn represent those passively gained from the mother. IgG is capable of fixing complement with order of efficacy being IgG3, IgG1 and IgG2. IgG4 cannot bind complement in the classical pathway. Only IgG3 and IgG 1 can bind to Fc receptors on macrophages.
IgM: This has high molecular weight and is also called as macroglobulin due to its large size. IgM molecules have a pentameric structure (i.e. five immunoglobulin units joined together) and also have an additional short polypeptide chain (J or joining chain). It comprises 5–10% of circulating immunoglobulins. IgM is the first antibody produced in response to the antigen (primary response). In contrast to IgG, IgM cannot cross the placenta. The foetus is able to produce IgM after maturation of its immune system. IgM is highly efficient in binding complement. A single molecule of IgM can bind complement while two molecules of IgG (lgG doublets) are necessary for complement-binding. The order of efficiency of complement binding of immunoglobulins is IgM, IgG3, IgG1 and IgG2. There are no receptors on macrophages for IgM.
IgA: There are two subclasses of IgA: IgA1 and IgA2. IgA is present mostly in body secretions such as gastrointestinal and respiratory mucosal secretions, saliva, tears, etc. Secretory IgA is mostly IgA2 and exists as a dimer. Serum IgA, which is mostly IgA1, is a monomer.
IgD and IgE: Both are present in trace amounts in serum and are monomeric.
Most IgD is expressed on the surface of resting B lymphocytes where it serves as an antigen receptor.
Most IgE is bound to basophils or mast cells through heavy chain. When a specific antigen combines with IgE, vasoactive substances are released from these cells and lead to anaphylaxis.
Alloantibodies vs autoantibodies: Alloantibodies are those which are produced by an individual against antigens present in another individual of the same species. Autoantibodies are those, which are produced by an individual against one's own antigens.
Warm vs cold antibodies: Warm antibodies react maximally at 37°C while cold antibodies show maximum activity at 0 to 4°C. Most IgG antibodies are of warm type while most IgM antibodies are of cold type. Characteristic features of different immunoglobulins are presented in table 1.3.
|
Complement
Complement are serum proteins which when activated react in an orderly manner with each other to cause immunologic destruction of target cells (lysis or phagocytosis). There are three pathways of complement activation: classical, alternate and mannose-binding pathway (Fig. 1.24).
Classical Pathway
Classical pathway is usually initiated by reaction of antibody (IgG or IgM) with antigen (e.g. red cells). Binding of only a single IgM pentameric molecule or of IgG doublet to an antigen are necessary for complement activation.
The complements are activated in the following order: Ag-Ab complex—C1 C4 C2 C3 C5 C6 C7 C8 C9. This process occurs on the surface of target cells (e.g. red cells). Binding of antibody to antigen causes exposure of complement-binding site on immunoglobulin. The activated C1 cleaves C4 to form C4a and C4b; C4a is released into the body fluid while C4b attaches to the red cell membrane. Activated C1 also cleaves C2 to form C2a. The C4b2a complex (C3 convertase) is formed. The C4b2a complex attached to cell membrane has enzymatic activity and can cleave several hundred C3 molecules. The C3a is released into plasma while C3b attaches to the cell membrane. C3b however is rapidly degraded into C3dg. C3b is not enzymatically active by itself, but presence of C3b on the cell surface is recognized by specific receptors on the surface of macrophages and this causes phagocytosis of C3b-bearing cells. C3dg cannot adhere to macrophages because macrophages do not have receptors for C3dg. Once C3b is converted to C3dg, then complement cascade is terminated; C3dg coated red cells in circulation are resistant to further complement-mediated cell destruction.
Some C3b joins C4b2a to form C4b2a3b (C5 convertase). C5 convertase cleaves C5 into C5a and C5b. C5a is released in circulation. C5b joins with C6 C7 C8 C9 to form membrane attack complex (MAC), which fixes on cell membranes and causes cell lysis. The MAC creates pores in red cell membrane through which water enters into red cells, cells swell and are lysed.

Figure 1.24: The complement pathway. Solid arrow indicates transformation of a complement component. Dashed arrow indicates enzymatic action of complement component that causes cleavage of that component
Alternate Pathway
In alternate pathway, C3 is activated directly with no role of earlier complement components. It does not require antigen-antibody reaction.
C3 can be activated by endotoxins, complex carbohydrates such as are present on some micro-organisms, and aggregates of IgA. A serum protein called properdin, factors B and D, and magnesium ions are needed for activation of alternate pathway.
Normally, C3 is being continuously cleaved at low level, probably by factor B, resulting C3b is rapidly cleared from the plasma. However, when C3b comes in contact with certain substances (e.g. complex carbohydrates on the surface of micro-organisms) then association of C3bB occurs on the surface of micro-organisms in the presence of Mg++ ions. Factor B is cleaved by factor D to form C3bBb. Properdin may stabilise C3bBb. C3bBb splits C3 to generate more C3b thus forming an amplification loop.
Alternate pathway plays an important role in initial defense against infection in nonimmune persons.
Mannose-binding lectin pathway: Mannose-binding lectin directly binds to target cell surface; this resembles binding of C1 to immune complexes and directly activates the classical pathway (without the need for immune complex formation).
Regulation of Complement Activity
Following factors act as a control mechanism against prolonged complement action:
- Specific inhibitors of activation of some complement components (particularly C1 and C3) are present in plasma.
- Enzymatically active complement components have a very short life and are rapidly degraded to inactive forms.
- Active fragments are rapidly cleared from circulation.
Various Effects of Complement Activation
- Opsonisation: Macrophages have specific receptors for C3b and thus target cells coated with C3b are recognised and phagocytosed by them (Opsonins are substances which when present on the surface of the antigen such as red cells facilitate immune phagocytosis; these are C3b and Fc portion of immunoglobulin which are recognized by specific receptors on the surface of macrophages).
- Target cell lysis by membrane attack complex C5b-9.
- Acute inflammation: Certain complement components play a role in acute inflammation. C3a and C5a are anaphylatoxins and increase vascular permeability. C5a, in addition, causes neutrophil chemotaxis.
MEGAKARYOPOIESIS
The process of development of megakaryocytes and platelets in bone marrow is known as megakaryopoiesis. It is divided into four stages (Fig. 1.25). Megakaryoblasts (stage I) are the earliest morphologically recognizable precursors; they are 6 to 24 μ in diameter, contain a single, large, oval, kidney-shaped, or lobed nucleus with loose chromatin and multiple nucleoli, and have deeply basophilic agranular cytoplasm. Promegakaryocytes (stage II) are larger than megakaryoblasts (15–30 μ), have lobulated or horseshoe-shaped nucleus, more abundant and less basophilic cytoplasm which may contain azurophil granules. Granular megakaryocytes (stage III) are 40 to 60 μ in diameter, contain a large multilobed nucleus with coarsely granular chromatin, and have abundant mildly basophilic cytoplasm containing numerous azurophil granules. Mature megakaryocytes (stage IV) are of similar size, contain a tightly packed multilobed and pyknotic nucleus, and have acidophilic cytoplasm; granules are arranged as ‘platelet fields’ (groups of 10–12 azurophil granules). Sometimes neutrophils or other marrow cells are seen traversing through the cytoplasm (emperipolesis); it has no clinical significance.
Mature megakaryocytes extend long and slender cytoplasmic processes (proplatelets) between endothelial cells of sinusoids in bone marrow and platelets are released from fragmentation of these processes. Each meakaryocyte produces 1000 to 5000 platelets, leaving behind a ‘bare’ nucleus which is removed by macrophages.
A unique feature of thrombocytopoiesis is endomitosis. This refers to nuclear division with cytoplasmic maturation but without cell division. As the cell matures from megakaryoblast to the megakaryocyte, there is gradual increase in cell size, number of nuclear lobes, and red-pink granules and gradual decrease in cytoplasmic basophilia. Megakaryocytes, the most abundant cells of the platelet series in the marrow, are large and contain numerous nuclear lobes with dense nuclear chromatin, and small aggregates of granules in the cytoplasm. The megakaryocytes possess well-developed membrane demarcation system.


Upon complete maturation, megakaryocytes extend pseudopods through the walls of the marrow sinusoids and individual platelets break-off into the peripheral circulation (Fig. 1.26). There is evidence that some of the megakaryocytes are carried to the lungs where platelets are released. A humoral factor, thrombopoietin, controls the maturation of megakaryocytes.
NORMAL HAEMOSTASIS
Haemostasis is the mechanism by which loss of blood from the vascular system is controlled by a complex interaction of vessel wall, platelets, and plasma proteins. Following vessel injury, haemostasis can be considered as occurring in two stages: primary and secondary. Primary haemostasis is the initial stage during which vascular wall and platelets interact to limit the blood loss from damaged vessel. During secondary haemostasis, a stable fibrin clot is formed from coagulation factors by enzymatic reactions. Although formation of blood clot is necessary to arrest blood loss, ultimately blood clot needs to be dissolved to resume the normal blood flow. The process of dissolution of blood clot is called as fibrinolysis.
The roles of vascular wall, platelets, and plasma proteins in normal haemostasis are briefly outlined below.
Vascular Wall
Endothelial cells synthesise certain substances which have inhibitory influence on haemostasis. These include—thrombomodulin, protein S, heparin-related substances, prostacycline (PGI2), and tissue plasminogen activator (tPA). Binding of thrombomodulin to thrombin causes activation of protein C. Protein C inactivates factors V and VIII: C and is a potent inhibitor of coagulation. Protein S is a cofactor for protein C. Deficiency of protein C or protein S is associated with tendency towards thrombosis. Heparin-like substances on the surface of endothelial cells potentiate the action of antithrombin. Prostacycline, a prostaglandin synthesised by endothelial cells, induces vasodilatation and also inhibits platelet aggregation. Endothelial cells also synthesise tissue plasminogen activator, which converts plasminogen to plasmin, and activates fibrinolytic system.
Certain factors synthesised by endothelial cells promote haemostasis and include tissue factor, von Willebrand factor and platelet activating factor. Tissue factor or thromboplastin activates extrinsic system of coagulation. von Willebrand factor mediates adhesion of platelets to subendothelium. Platelet activating factor induces aggregation of platelets (Fig. 1.27).
Another vascular factor promoting haemostasis is vasoconstriction of small vessels following injury.
Subendothelial collagen promotes platelet adhesion and also activates factor XII (intrinsic pathway).
Platelets
Platelets are derived from cytoplasmic fragmentation of bone marrow cells called megakaryocytes. They measure 2 to 3 μ in diameter. Normal platelet count in peripheral blood is 1.5 to 4 lacs/cmm. Platelets remain viable in circulation for approximately 10 days. About one-third of the total platelets in the body are in the spleen and remainder in peripheral blood. Under light microscope, in peripheral blood smears stained with one of the Romanowsky stains, platelets appear as small, irregular with fine cytoplasmic processes. Cytoplasmic granules are often visible. These granules may be packed in the central portion (granulomere) with peripheral cytoplasm appearing clear (hyalomere).

Ultrastructure of Platelets
Ultrastructurally, following three zones can be distinguished: (1) Peripheral zone: exterior coat (glycocalyx), cell membrane, open canalicular system; (2) Sol-gel zone: microfilaments, circumferential microtubules, dense tubular system; (3) Organelle zone: alpha granules, dense granules, mitochondria, lysosomes (Fig. 1.28).
Peripheral zone: Exterior or surface coat (glycocalyx) overlies the cell membrane. It is made of proteins, glycoproteins, and mucopolysaccharides. Some of the glycoproteins are polysaccharide side chains of the integral membrane proteins while others are adsorbed from the plasma.
The cell membrane is a trilaminar membrane composed of proteins, lipids, and carbohydrates. The chief membrane lipids are phospholipids which are arranged as a bilayer; the polar head groups are oriented both externally (towards plasma) and internally (towards cytoplasm) while the fatty acid chains are oriented toward each other. Phospholipids are distributed asymmetrically in the membrane with phosphatidylinositol concentrated on the inner half of the bilayer and phosphatidylethanolamine on the outer half. The phospholipids play an important role in prostaglandin synthesis and in platelet procoagulant activity.
An extensive open canalicular system, formed by invagination of the cell membrane, communicates with the exterior. It functions as a route through which platelet contents are secreted outside the cell.
Sol-gel zone: Microtubules provide structural support to the platelets. Microfilaments have contractile function. The dense tubular system, derived from smooth endoplasmic reticulum, is the site of pooling of calcium and formation of prostaglandin and thromboxane.

|
Organelle zone: Platelet organelles are alpha granules, dense granules, lysosomes and peroxisomes. Contents of platelet organelles are shown in Table 1.4.
Platelet Membrane Glycoproteins
The cell membrane contains integral membrane glycoproteins (Gp), which play an important role in haemostasis. Important platelet membrane glycoproteins and their functions are as follows:
Gp Ib-IX-V: This is a constitutively active receptor that mediates vWF-dependent adhesion of platelets to subendothelial collagen.
Gp IIb/IIIa: On activation, serves to bind fibrinogen and thus mediates aggregation. Also receptor for vWF, fibronectin, and thrombospondin.
Gp Ia-IIa: Constitutively active receptor for collagen and mediates platelet adhesion independent of vWF.
Platelet Antigens
Platelets possess HLA antigens and platelet-specific antigens. HLA class I antigens induce alloimmunisation and cause refractoriness to platelet transfusions when platelets are obtained from random donors. The platelet-specific antigen systems are now known as human platelet antigen (HPA) systems. Platelet specific antigens play an important role in neonatal alloimmune thrombocytopaenic purpura (NATP) and in post transfusion purpura.
Role of Platelets in Haemostasis
Activation of platelets refers to adhesion, aggregation, and release reaction of platelets which occurs after platelet stimulation (i.e. after vascular damage).
Adhesion: This means binding of platelets to nonendothelial surfaces, particularly subendothelium which is uncovered following vascular injury. von Willebrand factor (vWF) mediates adhesion of platelets to subendothelium via GpIb on the surface of platelets (Fig. 1.29). Congenital absence of glycoprotein receptor GpIb (Bernard-Soulier syndrome) or of von Willebrand factor in plasma (von Willebrand's disease) causes defective platelet adhesion and bleeding disorder.

Figure 1.29: Platelet adhesion to subendothelial collagen. GpIb receptor on platelets and von Willebrand factor are necessary for attachment of platelets to subendothelial collagen
Platelets normally circulate as round to oval disc-like structures. With activation, platelets undergo shape change, i.e. they become more spherical and form pseudopodia.
This shape change is due to reorganisation of microtubules and contraction of actomyosin of microfilaments.
Release reaction (secretion): Immediately after adhesion and shape change, process of release reaction or secretion begins. In this process, contents of platelet organelles are released to the exterior. ADP released from dense granules promotes platelet aggregation. Platelet factor 4 released from alpha granules neutralises the anticoagulant activity of heparin while platelet-derived growth factor stimulates proliferation of vascular smooth muscle cells and skin fibroblasts and plays a role in wound healing.
Activated platelets also synthesise and secrete thromboxane A2 (TxA2) (Fig. 1.30). Platelet agonists such as ADP, epinephrine, and low-dose thrombin bind to their specific receptors on platelet surface, and activate phospholipase enzymes, which release arachidonic acid from membrane phospholipids. Arachidonic acid is converted to cyclic endoperoxides PGG2 and PGH2 by the enzyme cyclo-oxygenase. These are then converted to thromboxane A2 by thromboxane synthetase. Thromboxane A2 has a very short half-life and is degraded into thromboxane B2 which is biologically inactive. TxA2 causes shape change and stimulates release reaction from alpha and dense granules. TxA2 also induces aggregation of other platelets and local vasoconstriction.
Aggregation: This may be defined as binding of platelets to each other. ADP released from platelets or from damaged cells binds to specific receptors on platelet surface. This causes inhibition of adenyl cyclase and reduction in the level of cyclic AMP in platelets. A configurational change in the membrane occurs so that receptors for fibrinogen (GpIIb and IIIa) become exposed on the surface. Binding of fibrinogen molecules to GPIIb/IIIa receptors on adjacent platelets causes platelet aggregation (Fig. 1.31).

Figure 1.30: Synthesis of thromboxane A2. Modes of action of certain antiplatelet drugs are also shown

Figure 1.31: Platelet aggregation. This requires binding of fibrinogen molecules to GpIIb/IIIa receptors on platelets
The activated platelets release ADP and TxA2 and so a self-sustaining reaction is generated leading to the formation of a platelet plug. Thrombin generated from activation of coagulation system is a potent platelet-aggregating agent and also converts fibrinogen to fibrin. Fibrin and aggregated mass of platelets at the site of injury constitute the haemostatic plug.
Platelet procoagulant activity: When platelets are activated, negatively charged phospholipids (phosphotidylserine and phosphatidylinositol) located in the inner half of the lipid bilayer become exposed on the outer surface. These phospholipids play active role in coagulation by providing surface for interaction of some coagulation factors. Critical coagulation reactions for which activated platelets provide a negatively charged phospholipid (PL) surface are shown in Figure 1.32.
Platelets may play a role in the activation of F XII in the presence of ADP and kallikrein. Platelets also can directly activate F XI independent of F XII. This may explain the absence of bleeding diathesis in persons with F XII deficiency.

Figure 1.32: Platelet procoagulant activity. Platelets provide surface for some important coagulation reactions
In addition platelets also secrete calcium, FV, fibrinogen, and FXII and contribute to the coagulation system.
Plasma Proteins in Haemostasis
Plasma proteins in haemostasis can be divided into following groups:
- Coagulation system: Factors I, II, III, IV, V, VII, VIII, IX, X, XI, XII, XIII, prekallikrein, high molecular weight kininogen.
- Fibrinolytic system: Plasminogen, plasmin, tPA, α2- antiplasmin, PAI-1, PAI-2.
- Inhibitor system: Protein C, protein S, antithrombin.
Coagulation System
A number of coagulation proteins (factors) participate in coagulation reactions, which ultimately lead to the formation of a fibrin clot. According to the International System of Nomenclature, coagulation factors are designated by Roman numerals (I to XIII). Table 1.5 lists the blood coagulation factors; common names and synonyms are given on the right side. Coagulation proteins can be divided into following categories: (1) Fibrinogen (F I); (2) Serine proteases: (a) Vitamin K-dependent Factors—II, VII, IX, X, (b) Contact factors—XI, XII, high molecular weight kininogen, prekallikrein; (3) Cofactors—V, VIII, tissue factor (F III); and (4) Transglutaminase: F XIII.
The coagulation factors have been assigned Roman numerals according to the order of their discovery. Except calcium and thromboplastin, all the coagulation factors listed in Table 1.5 are glycoproteins. When coagulation factors become activated, they are converted from an inactive zymogen form to a serine protease. However, factors V and VIII, when activated, do not develop enzymatic activity but become modified and are called as cofactors; in their absence the reactions, which they modify, become markedly slow. Activation of fibrinogen denotes cleavage of fibrinopeptides A and B from the molecule with formation of fibrin. Factors II, VII, IX, and X are called as vitamin K-dependent factors. Vitamin K is required for γ-carboxylation of these proteins, which is necessary for calcium binding. Calcium in turn, is necessary for binding of these coagulation factors to phospholipid surface. Attachment of coagulation factors to phospholipid is essential for coagulation reactions to occur. In the absence of vitamin K, carboxylation fails to occur and functionally inactive forms of vitamin K-dependent factors are produced. Factors XII, XI, high molecular weight kininogen, and prekallikrein are called as contact factors; they are involved in the activation of coagulation via intrinsic pathway.
|
The liver is the site of synthesis of most coagulation factors. However, F XIII is derived from megakaryocytes, while vascular endothelial cells and megakaryocytes synthesize von Willebrand factor.
Individual coagulation factors are considered briefly below:
Fibrinogen (Molecular weight (MW) 340,000; 1/2 life 90 hours): The level of fibrinogen in plasma is the greatest among the coagulation proteins and ranges from 200 to 400 mg/dl. Fibrinogen molecule consists of three pairs of polypeptide chains Aα, Bβ, and γ which are held together by disulfide bonds. The complete molecule is represented as Aα2 Bβ2 γ2. Fibrinogen consists of three domains—two outer D domains and a central E domain. Fibrinopeptides A and B are located in the central domain at the N-terminals of Aα and Bβ chains, respectively. Thrombin releases fibrinopeptides A and B from these chains to form fibrin monomers (Fig. 1.38). Fibrinogen is an acute phase reactant and its concentration rises in a variety of non-specific conditions such as inflammation, trauma, and myocardial infarction.
Prothrombin (MW 72,000; 1/2 life 60 hours): Factor II or prothrombin is converted to thrombin by the enzyme complex Xa-V-phospholipid-calcium (called as prothrombinase). Thrombin has multiple functions in haemostasis (Fig. 1.33).
- Thrombin splits fibrinopeptides A and B from fibrinogen to form fibrin monomers;
- Thrombin activates F XIII which is necessary for cross-linking of fibrin and stabilisation of the clot.
- Thrombin activates Factors VIII and V which in turn enhance the activation of F X and prothrombin, respectively.
- Thrombin is a powerful platelet agonist.
- Thrombin activates protein C, a natural anticoagulant.

Thromboplastin: Tissue factor is required for activation of F VII in extrinsic pathway. It is composed of two parts: protein and phospholipid. Tissue factor is distributed in all tissues, but especially high concentrations are present in brain, placenta, and lungs.
Factor V (MW 330,000; 1/2 life 12–36 hours): F V is a heat-labile factor, which is inactivated rapidly at room temperature in vitro. F V is activated by thrombin and functions as a cofactor in the conversion of prothrombin to thrombin by the prothrombinase complex. About one-fifth of the FV in blood is stored in platelet alpha granules, which is released when platelets are activated.
Factor VII (MW 48,000; 1/2 life 6 hours): Tissue injury results in the formation of a complex between single chain form of F VII, tissue factor, and calcium which generates small amount of F Xa from F X. Factor Xa then in the reverse reaction cleaves F VII to yield F VIIa (two chain form); the F VIIa- tissue factor-calcium complex has greatly increased activity. This complex can also activate F IX.
Factor VIII (MW of F VIII: C-200,000; 1/2 life of F VIII: C approx 12 hours): The F VIII circulates in plasma as a noncovalently bound complex of two components—F VIII:C and von Willebrand factor. F VIII:C is the low molecular weight portion which has procoagulant activity and its synthesis is X-linked. F VIII mRNA is detected in various tissues; liver however, appears to be the primary source of F VIII. von Willebrand factor is the high molecular weight component which is autosomal in inheritance and synthesised by endothelial cells and megakaryocytes (Fig. 1.34). von Willebrand factor functions as a carrier protein for F VIII:C and also mediates adhesion of platelets to the subendothelium at sites of vessel damage.
The gene that codes for F VIII is located on the long arm of the X chromosome. It is 186 kilobases long and consists of 26 exons. The RNA is approximately 9 kilobases in length. The F VIII protein is composed of various domains, which are arranged as A1-A2-B-A3-C1-C2. A1 and A2 domains constitute the heavy chain of the molecule while A3, C1, and C2 make up the light chain. B is the connecting region (Fig. 1.35).


Figure 1.35: Factor VIII gene and factor VIII molecule. F VIII gene has 26 exons and 25 introns. Polypeptide chain encoded by the gene has six regions: A1, A2, B, A3, C1, and C2. Proteolytic action by thrombin (small arrows) produces activated F VIII molecule
Thrombin proteolytically cleaves F VIII molecule at three different positions to form activated F VIII. Activated F VIII functions as a cofactor in the reaction F X → FXa and enhances the velocity of this reaction several thousand-fold. Further cleavage by thrombin and activated protein C inactivates VIII.
Various terms and definitions related to F VIII and von Willebrand factor are given below:
- Vlll vWF: Complex of FVIII procoagulant protein and von Willebrand factor
- F VIII: C: F VIII coagulant activity which is measured by clotting assay
- F VIII: C Ag: Antigenic expression of F VIII measured by immunologic technique
- VWF: A multimeric protein necessary for platelet adhesion
- VWF:RCo: Ristocetin cofactor activity, the activity of von Willebrand factor required for ristocetin-induced platelet aggregation
- VWF Ag: Antigenic expression of von Willebrand factor measured by immunological technique.
Factor IX (MW 57,000; 1/2 life 24 hours): F IX, a vitamin K-dependent glycoprotein, is activated by F XIa or by F VIIa-tissue factor complex to F IXa, a two-chain molecule. F IX is inherited in a sex-linked manner.
Factor X (MW 58,000; 1/2 life 20–40 hours): F X, a vitamin K-dependent protein, is activated by both intrinsic (i.e. F IXa-VIII-phospholipid-calcium complex) and extrinsic (i.e. tissue factor-VII complex) pathways. It is necessary for the formation of prothrombinase (Xa-V-phospholipid-calcium) in the common pathway.
Factor XI (MW 160,000; 1/2 life 40–80 hours): F XI is activated by F XIIa in the presence of high molecular weight kininogen. Its activity increases upon storage.
Factor XII (MW 80,000; 1/2 life 40–50 hours): F XII is activated when it comes in contact with substances such as collagen, glass, celite, ellagic acid, etc. F XIIa converts F XI to its active form and also prekallikrein to kallikrein. F XII plays a role in contact activation of coagulation system, inflammatory response, complement system, fibrinolysis, and formation of kallikrein and kinin.
Factor XIII (MW 320,000; 1/2 life 3–7 days): In contrast to all other coagulation factors, F XIII is a transglutaminase. Activated F XIII catalyzes the formation of covalent bonds between adjacent molecules of fibrin monomer (cross-linking) which provides stability to the fibrin clot.
Prekallikrein (MW 88,000; 1/2 life 35 hours): Prekallikrein is activated by F XIIa to kallikrein. Kallikrein in turn further activates F XII and thus serves to amplify the initial stimulus. Kallikrein plays a role in chemotaxis and in activation of fibrinolysis. Kallikrein also converts high molecular weight kininogen to bradykinin, a chemical mediator of inflammation.
High molecular weight kininogen (MW 110,000; 1/2 life 6.5 days): This circulates in plasma complexed to pre-kallikrein and F XI. It promotes contact activation.
Mechanism of Blood Coagulation
Scheme of blood coagulation is divided into intrinsic, extrinsic, and common pathways (Fig. 1.36). The intrinsic pathway is initiated by contact activation and consists of interaction of contact factors (F XII, F XI, prekallikrein, and high molecular weight kininogen), F IX, F VIII, phospholipid, and calcium; these reactions generate a complex which causes activation of F X to F Xa.

Figure 1.36: Scheme of blood coagulation. Solid arrows indicate transformation. Broken lines indicate action. Abbreviations: HMWK: High molecular weight kininogen; TF: Tissue factor; PL: Phospholipid; Ca++: Calcium
The extrinsic pathway is initiated by tissue injury with release of tissue thromboplastin which causes activation of F VII; the enzyme which is formed activates F X. Both intrinsic and extrinsic pathways proceed to common pathway which begins with activation of F X, involves interaction of F X, F V, prothrombin, phospholipid, calcium, and F XIII and leads to the formation of fibrin.
Intrinsic pathway: Initiation of intrinsic pathway occurs when plasma comes in contact with a negatively charged surface such as glass, kaolin, celite, or ellagic acid in vitro. In vivo, this surface is probably provided by subendothelium of a damaged vessel. Following contact with a negatively charged surface, a conformational change in FXII with exposure of enzymatically active site probably occurs and in this way a small amount of F XIIa is formed. F XIIa converts prekallikrein to kallikrein and F XI to FXIa in the presence of high molecular weight kininogen. Kallikrein in turn activates more F XII thus providing autoamplification of the reaction. F XIa cleaves F IX to yield F IXa; this reaction requires the presence of phospholipid and calcium. F IXa complexes with activated F VIII, phospholipid, and calcium and activates F X to F Xa. F VIII is activated by thrombin and also by F Xa. F VIII does not possess enzymatic activity but functions as a cofactor; in its presence the reaction rate is enhanced several thousand times.
Extrinsic pathway: F VII complexes with tissue factor released after tissue injury in the presence of calcium ions and activates F X and F IX. F Xa and thrombin convert 45the single-chain form of F VII to the two-chain form, which has greatly increased enzymatic activity as compared to the single-chain form. This reciprocal activation of F VII leads to autoamplification of the reaction.
The concept of intrinsic and extrinsic pathways of blood coagulation is applicable to in vitro blood clotting. It is uncertain whether intrinsic pathway plays any significant role in vivo. This is because of observed absence of haemorrhagic tendencies in patients of F XII, prekallikrein, or HMWK deficiency. Also, in addition to F VII of extrinsic pathway, tissue factor has also been shown to activate F IX to F IXa in intrinsic pathway. In vivo, blood clotting seems to be initiated primarily by tissue factor.
Common pathway: Common pathway begins with the activation of F X. F Xa generated by intrinsic or extrinsic pathway complexes with F V, phospholipid and calcium. This is called as prothrombinase complex, which activates prothrombin to thrombin. FV is modified by thrombin or F Xa to form activated F V which functions as a cofactor in the above reaction. Thrombin removes fibrinopeptides A and B from α and β chains of the fibrinogen molecule to form fibrin monomer. Free fibrin monomers spontaneously polymerize by forming end-to-end and side-to-side non-covalent bonds with each other. This is called as fibrin polymer. F XIIIa (generated from F XIII by thrombin), in the presence of calcium, mediates the formation of covalent bonds between adjacent polypeptide chains. This cross-linking of fibrin monomers imparts structural stability to the clot (Fig. 1.38).
Physiologic mechanism of coagulation: Under physiologic conditions in vivo, tissue factor (TF) and F VIIa initiate coagulation. F VIIa-TF complex activates F IX to F IXa, which then activates F X to F Xa. F Xa in turn activates prothrombin to thrombin, which converts fibrinogen to fibrin. If stimulus for thrombin formation is strong enough, coagulation is maintained through the activation of F XI by thrombin. Thrombin also activates F VIII, F V, and F XIII (Fig. 1.37).
Fibrinolytic System
Fibrinolysis is the process of dissolution of blood clots which is necessary to maintain the free flow of blood in the vascular system. The major enzyme of the fibrinolytic system is plasmin, which is generated from proteolytic cleavage of plasminogen. Plasmin can cause cleavage of both fibrinogen as well as fibrin. Plasmin digests insoluble or cross-linked fibrin to release fibrin degradation products or FDPs which are then cleared from the circulation by macrophages of the mononuclear phagocytic system.
Plasminogen is converted to plasmin by plasminogen activators. This reaction occurs on the surface of fibrin. The plasminogen activators include—(1) tissue plasminogen activator (tPA): This is synthesized by endothelial cells and is the most important physiological plasminogen activator. tPA most efficiently converts plasminogen to plasmin when plasminogen is bound to the fibrin clot; (2) kallikrein (formed from prekallikrein by the action of F XIIa) converts plasminogen to plasmin (Fig. 1.39).

Figure 1.37: Physiologic mechanism of coagulation: Under physiologic conditions in vivo, tissue factor (TF) and F VIIa initiate coagulation. F VIIa-TF complex activates F IX to F IXa, which then activates F X to F Xa. F Xa in turn activates prothrombin to thrombin, which converts fibrinogen to fibrin. If stimulus for thrombin formation is strong enough, coagulation is maintained through the activation of F XI by thrombin. Thrombin also activates F VIII, F V, and F XIII and plays a central role in coagulation
Inhibitors of fibrinolysis: These include (1) α2-antiplasmin which combines rapidly with plasmin in circulation to form plasmin-antiplasmin complex; (2) α2-macroglobulin which inhibits plasmin; (3) plasminogen activator inhibitors PAI-1 and PAI-2 released from endothelial cells which neutralize tPA and (4) thrombin-activated fibrinolytic inhibitor (TAFI) which cleaves specific fibrin lysine residues, thus removing binding sites for plasminogen and tPA.
Fibrinogen degradation products: Plasmin initially attacks α chains of the fibrinogen molecule and removes small fragments designated as A, B, and C from the C-terminals of the Aα chains. This is followed by degradation of Bβ chains with removal of first 42 amino acids. This leads to the formation of a large fragment X that still retains fibrinopeptide A. The next cleavage involves all the three chains in an asymmetrical manner with the release of fragment Y and fragment D. Fragment Y is rapidly degraded by plasmin liberating two fragments D and E (Fig. 1.40).
Fibrin degradation products: Degradation of cross-linked fibrin is different from that of fibrinogen. Firstly, the fibrin degradation products are different because of the presence of covalent bonding. Thus, the characteristic fragments are oligomers of X and Y, D-dimer, D2E complex, and Y-D complex. Secondly, fibrin degradation is slower due to the presence of cross-linkages (Fig. 1.41)
Effects of FDPs: Normally, the FDPs are cleared from the circulation by macrophages of the reticuloendothelial system. However, when FDPs increase they have a potent anticoagulant action in the form of inhibition of polymerisation of fibrin, antithrombin activity, and impairment of platelet function.

Figure 1.38: Steps in the formation of cross-linked fibrin. 1 and 2: Cleaving of fibrinopeptides from fibrinogen by thrombin to form fibrin monomers. 3: Spontaneous polymerisation of fibrin monomers. 4: Cross-linking of fibrin monomers mediated by F XIIIa. During this stage, covalent bonds form mainly between adjacent γ chains. For simplicity Aα chains are not shown


Natural Inhibitors of Coagulation
There are three main physiologic inhibitors of coagulation that are present in normal plasma. These are antithrombin (previously called antithrombin III), protein C and protein S system, and tissue factor pathway inhibitor (Fig. 1.42).
Antithrombin (AT): AT is the most important physiologic inhibitor of coagulation. This is a single chain glycoprotein synthesized by the liver. AT possesses inhibitory activity principally against thrombin and to a lesser extent against factors Xa, XIa, XIIa, and IXa. AT binds with thrombin and other serine proteases to form a stable complex. Heparin-like substances present on the luminal surface of blood vessels promote activity of AT. The importance of AT as a natural anticoagulant derives from the fact that AT deficiency is associated with increased risk of thrombosis.
Heparin binds with AT and potentiates its action. This is the basis of efficacy of heparin as a therapeutic anticoagulant.
Protein C: Protein C is a vitamin K-dependent glycoprotein synthesized in the liver. Protein C circulates in an inert zymogen form and is activated by thrombin in the presence of thrombomodulin on the surface of vascular endothelial cells.


Figure 1.42: Natural inhibitors of coagulation: These include (1) tissue factor pathway inhibitor (TFPI) (binds to F Xa and then to VIIa-TF-Ca++ complex), (2) antithrombin (AT) (inhibits mainly thrombin and F Xa) and (3) protein C pathway (inactivates activated forms of F VIII and F V)
50Protein C causes proteolytic destruction of activated factors V and VIII. Protein S, another vitamin K-dependent protein, functions as a cofactor in this reaction and enhances the action of protein C. Protein C also appears to enhance fibrinolysis.
An inhibitor of protein C is present in plasma; it is thought that deficiency of this inhibitor accounts for cases of combined deficiency of F V and FVIII.
Deficiency of protein C or S is associated with risk of thrombosis.

Tissue factor pathway inhibitor (TFPI): This binds to FXa, and FXa-TFPI complex then attaches to tissue factor-VII complex to neutralize it.
BIBLIOGRAPHY
- Baugh RF, Hougie C Structure and function in blood coagulation. In Poller L (Ed): Recent Advances in Blood Coagulation. No. 3. Churchill Livingstone. Edinburgh . 1981.
- Bottomley SS, Muller-Eberhard U Pathophysiology of heme synthesis. Semin Hematol. 1988; 25: 282–302.
- Brommer EJP, Brakman P Developments in fibrinolysis. In Poller L (Ed): Recent Advances in Blood Coagulation. No. 5. Churchill Livingstone, Edinburgh . 1991.
- Cannistra SA, Griffin JD Regulation of the production and function of granulocytes and monocytes. Semin Hematol. 1988; 25: 173–88.
- Collen D, Lijnen HR Basic and clinical aspects of fibrinolysis and thrombolysis. Blood. 1991; 78: 3114–24.
- Dacie JV, Lewis SM Practical Haematology. 7th ed. Churchill Livingstone. Edinburgh . 1991.
- Furie B, Furie BC Molecular and cellular biology of blood coagulation. N Engl J Med. 1992; 326: 800–806.
- Gerrard JM, Didisheim P Platelet structure, biochemistry and physiology. In Poller L (Ed): Recent Advances in Blood Coagulation. No. 3. Churchill Livingstone. Edinburgh . 1981.
- Greer JP, Foerster J, Rodgers GM, Paraskevas F, Glader B, Arber DA, Means RT Jr (Eds): Wintrobe's clinical hematology. 12th ed. Lippincott Williams & Wilkins. Philadelphia . 2009.
- Groopman JE Colony-stimulating factors: Present status and future applications. Semin Hematol. 1988; 25: 30–37.
- High KA, Benz EJ The ABC of molecular genetics. A haematologist's introduction. In Hoffbrand, AV (Ed): Recent Advances in Haematology Vol. 4. Churchill Livingstone. Edinburgh . 1985.
- Holmsen H Platelet metabolism and activation. Semin Hematol. 1985; 22: 219–40.
- Johnston RB Monocytes and macrophages. N Engl J Med. 1988; 318: 747–752.
- Kumar V, Abbas AK, Fausto N, Aster JC: Robbins and Cotran Pathologic basis of disease. 8th Ed. Saunders Elsevier. Philadelphia . 2010.
- Lenting PJ, van Mourik JA, Mertens K The life cycle of coagulation factor VIII in view of its structure and function. Blood. 1998; 92: 3983–96.
- McPherson RA, Pincus MR (Eds): Henry's clinical diagnosis and management by laboratory methods. 21st ed. Elsevier Saunders. Philadelphia . 2007.
- Mohandas N, Chasis JA Red blood cell deformability, membrane material properties and shape: Regulation by transmembrane, skeletal, and cytosolic proteins and lipids. Semin Hematol. 1993; 30: 171–92.
- Nathan CF Secretory products of macrophages. J Clin Invest. 1987; 79: 319–26.
- Ogston D, Bennett B Blood coagulation mechanism. In Poller L (Ed): Recent Advances in Blood Coaguation. No. 5. Churchill Livingstone. Edinburgh . 1991.
- Parslow TG Immunoglobulin genes, B cells, and the humoral immune response. In Stites DP, Terr AL, Parslow TG (Eds): Basic and Clinical Immunology. 8th ed. Appleton and Lange. Connecticut . 1994.
- Sixma JJ The haemostatic plug. In Poller L (Ed): Recent Advances in Blood Coagulation. No. 3. Churchill Livingstone. Edinburgh . 1981.
- Spangrude GJ Biologic and clinical aspects of hematopoietic stem cells. Annu Rev Med. 1994; 45: 93–104.
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (Eds): WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. International Agency for Research on Cancer. Lyon . 2008.
- Van der Valk P, Herman CJ Leucocyte functions. Lab Invest. 1987; 56: 127–37.
- Williams D and Nathan DG Introduction: The molecular biology of hematopoiesis. Semin Hematol. 1991; 28: 114–116.