

INTRODUCTION
Contrary to the pervasive assumption, bones are not just inert structures which hold the body in erect position, facilitate locomotion, or house various organs in the body. Instead, skeletal tissue is a dynamic organ and remains metabolically active throughout the life, participating in mineral homeostasis and maintenance of body infrastructure. Bones serve three principle functions—mechanical support for the body, sites for muscle and tendon insertions for locomotion, and as reservoirs for calcium and phosphate for the organism. To the extent it is effective, bone remodeling helps maintain vitality and integrity of the entire skeleton for the life of a person, without which we would crumble to the ground sooner rather than later.1,2
BONE STRUCTURE AND DISTRIBUTION
Human skeleton consists of 80% cortical and 20% cancellous bone; the former is mainly in the appendicular skeleton providing stature and serve locomotive function and the latter mainly in the axial skeleton primarily serving hematopoietic and mineral homeostatic functions. Both types of bone serve as reservoirs for calcium and to a lesser extent phosphate. Macroscopically, all bones in the body consist of two distinct compartments; an outer cortex or compacta, a more solid structure, surrounding trabecular bone consisting of interconnected plates and bars of about 200 µm thickness distributed in three dimensions. This unique arrangement results in three surfaces in cortical bone: A periosteal surface on the outside, an endosteal or endocortical surface on the inner surface of the cortex, and an intracortical surface inside the Haversian system (Fig. 1). In the trabecular bone, there are only two surfaces—trabecular and intratrabecular with a smaller proportion of periosteocytic lacunar surface (Fig. 1). Each of these surfaces is always in one of the three functional states—forming surfaces covered by cuboidal osteoblasts, resorbing surfaces covered by osteoclasts, and quiescent surfaces covered by thin layer of flat lining cells, sometimes referred to as resting osteoblasts or surface osteocytes (Fig. 2).1,2
Although trabecular bone accounts for only about 20% of the total body bone volume, it contributes almost 70% to the total body bone surface. Because of this high surface to volume ratio, the rate of remodeling in trabecular bone is about fourfold higher than in cortical bone. Any alteration in bone remodeling (physiologic, metabolic, or pathologic) has greater impact on trabecular than on cortical bone.32


FIG. 1: Structure of cortical and trabecular bone: Schematic depiction of cortical or compact bone that consists of a periosteal surface on the outside, an endosteal or endocortical surface on the inner surface of the cortex, and an intracortical surface inside the Haversian system. Shown also, cancellous or trabecular bone, a sponge-like structure formed by the interconnected trabecular plates.
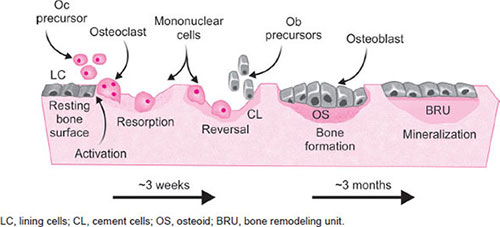
FIG. 2: Bone remodeling sequence: At discrete time and place, bone remodeling begins with osteoclastic resorption which lasts about 2 weeks. This is followed by a week of reversal phase in which preosteoblasts are recruited and differentiate into mature mononuclear cuboidal osteoblasts laying down osteoid. Mineralization of osteoid is rapid during the first 2–4 weeks, accounting for approximately 80% of the final degree of mineralization of bone followed by slow secondary mineralization (~20%) over the next 4–8 weeks or longer. Bone formation, which includes laying down osteoid and its subsequent mineralization takes approximately 3 months.
Similarly, any pharmacologic manipulation will have a greater effect on trabecular than on cortical bone. It is important to appreciate this unique behavior of the two bony compartments in health and disease.
Bones contain a large number of structural units adjacent to each other which are formed at different times, and separated by a thin layer of mineralized ground substance called cement line. The structural unit of cortical bone is the Haversian system or an3 osteon, a cylinder of concentric lamellae of ~200 µm in diameter, with its long axis roughly parallel to the long axis of the bone (Fig. 1). It has a central canal of about 60 µm in diameter containing blood vessels that are connected at the endosteal surface with those of the bone marrow. The structural unit of trabecular bone is conventionally referred to as trabecular packet. Both cortical and trabecular bone contain lacunae and canaliculi within which osteocytes and their cell processes reside, thus maintaining contact with each other and with the cells on the surface (Fig. 1). Except at sites of active remodeling, cortical and trabecular bone surfaces are separated from bone marrow by a thin layer of flat lining cells.
Microscopically, the structure of cortical and trabecular bone is similar and is composed of lamellar bone. In contrast, woven bone is found in early embryonic development, during fracture healing, and in various disease states such as Paget's disease of bone, hyperparathyroidism (primary or secondary), fibrous dysplasia, etc. In the normal bone, each lamella is about 2 µm in thickness and has a similar pattern of fiber orientation. At intervals between adjacent lamellae are lacunae containing osteocytes that interconnect with each other and with lining cells on bone surfaces with their long canaliculi. The osteocytes and associated canalicular network are analogous to the neuronal network in the nervous system. In contrast, woven bone has no regular pattern of collagen fiber orientation and the osteocyte lacunae are larger, more prevalent, and are randomly distributed, which gives rise to the characteristic mosaic appearance under polarized microscopy. The woven bone is best suited for rapid temporary filling of gaps, as occurs after a fracture, but it is biomechanically much weaker than the normal lamellar bone.
BONE CELLS AND FUNCTION
The principal cells of bone are osteoclasts, osteoblasts, and osteocytes (Fig. 3), which have striking similarity to hemopoietic cells—red cells, white cells, and platelets. Just as the hemopoietic cells serve different functions, so do bone cells. Osteoclasts (derived from hematopoietic progenitors), osteoblast (derived from bone marrow stromal cells with close genealogic relationship to adipocytes), and osteocytes (formed by transformation of osteoblasts after they have accomplished their task of laying down new matrix) are not only under the control of systemic hormonal influences, but also respond to local cytokine, growth factors, and biomechanical influences (Fig. 4). A full exposition of the detailed orchestration of this complex system is beyond the scope of current discussion. Interested readers are directed to the excellent reviews1,2 as well as to the listed websites.
Osteoclasts are multinucleated large cells which resorb bone, either old bone in normal physiologic conditions or normal bone in pathologic conditions (metabolic disorders or metastatic bone disease). Osteoclasts form a sealed compartment by attaching to the surface of bone destined to be renewed or removed with their characteristic ruffled border, referred to as Howships’ lacunae. Within this compartment osteoclasts secrete acid, mediated by carbonic anhydrase, to degrade bone, exactly the same process used to decalcify bone with acid for routine pathologic examination. Following resorption of a finite, presumably predefined area of old bone, osteoclasts undergo apoptosis releasing humoral agents that most likely trigger recruitment of osteoblast to refill the excavated bone surface.
Osteoblasts are mononuclear cuboidal or columnar cells and make proteins that form the organic matrix that is eventually mineralized to form mature normal lamellar bone structure. When a team of osteoblasts completed their task, referred to as wall thickness (a packet), many osteoblasts transform into osteocytes (Fig. 3) and are buried within the mineralized bone, and the rest undergo apoptosis.4

FIG. 3: Origin of principal cells involved in bone remodeling. The pluripotential mesenchymal stem cells is the origin of both osteoblasts and adipocytes. During differentiation the stem cell has to commit either to adipocyte or osteoblast lineage. At present the signaling mechanisms that control this pathway are unknown, but provide basis for the concept of fat-bone connection. After laying down matrix, the cuboidal osteoblasts will be incorporated into mineralized bone as osteocytes with its canalicular system connecting with each other and with lining cells on the bone surface. Osteoclasts are derived from myeloid precursors and transform from mononuclear preosteoclast to multinucleated osteoclasts. Both osteoblasts and osteoclast undergo apoptosis after they accomplished their task.
Osteocytes are, as far as is currently known, considered the direct descendants of osteoblasts (see above). Osteocytes reside in the osteocytic lacunae, and connect with each other and to the lining cells on the bone surface with their long canaliculi to form a network similar to the neuronal network. Until recently, the role of osteocytes in bone biology and mineral homeostasis was poorly understood and at one time thought to be inert cells with no known function. In the past decade, much has been learned about their role in bone health, bone integrity, and in phosphate regulation (see chapter on Disorders of Phosphorus Metabolism), an unexpected finding. The two most prominent secretions of osteocytes are sclerostin, a potent inhibitor of bone formation, and fibroblast growth factor-23 (FGF-23), involved in phosphate regulation.4,5
MODELING AND REMODELING
To be healthy, bones must renovate themselves on a continual basis throughout life. This is accomplished by bone remodeling, the cyclical replacement of old bone by new, which serves to maintain its mechanical and metabolic functions. In each cycle a circumscribed predetermined volume of bone is removed by osteoclastic resorption and subsequently, replaced by osteoblastic formation at the same location. Often bone remodeling is inappropriately used interchangeably with bone turnover; the former is a process by which it is renovated and the latter is the rate at which it is occurring.
In contrast, modeling occurs only during growth and development when bones are lengthening to attain a predetermined size and taking shape appropriate for the site in the body and site specific mechanical needs.4,6 There is emerging evidence that modeling can be induced by pharmacologic manipulation with anabolic agents,7 but the extent, magnitude, and durability of this process in preventing age related bone loss and preserving bone integrity is not fully explored.5

FIG. 4: Hormones and cytokines regulating bone cell function: Schematic representation of various factors regulating bone cell differentiation, function, and remodeling.
Bone remodeling is carried out by elongated structures known as basic multicellular units (BMUs) that travel through or across the surface. Each BMU lasts about 6 months with continued sequential recruitment of new osteoclasts and osteoblasts, removes a moiety of old bone and replaces with new bone of the same quality and quantity. Each remodeling cycle begins with bone resorption followed closely in space and time by bone formation, referred to as coupling, with prompt and complete refilling of the resorption cavity with new bone. However, this focal regulation of bone balance by “coupling phenomenon” is much more complex than is generally assumed.
A remodeling cycle consists of four successive stages: activation, resorption, reversal, and formation followed by quiescence until the next remodeling cycle begins when the newly laid down bone become old. In children, adolescents, and adults, bone remodeling is an efficient system with bone resorption and formation are coupled, and keep pace with each other regardless of rate of turnover (high, normal, or low). However, the system is perturbed by aging, hormonal influences, or disease processes, commonly referred to as uncoupling. It begins with recruitment of preosteoclasts and their functional expression, and recruitment of preosteoblasts and their functional expression. The initial signal for6 activation probably comes from osteocytes, which are mechanosensors buried deep within the bone matrix.8 The detailed signaling pathways to initiate the remodeling process are not fully understood and the subject is beyond the scope of the subject. Since, the remodeling process is not in tandem, all four processes are ongoing, simultaneously. In cortical bone, the BMU consists of a cutting cone of osteoclasts resorbing an old moiety of bone, a quiescent zone, and a closing cone of osteoblasts laying down new young matrix, which eventually mineralizes the entire complex advancing through the bone to build a new Haversian system. This is somewhat analogous to digging tunnels to build roads through mountains or underneath water ways with tunneling machines that dig the earth at the advancing front followed immediately behind by building the tunnel structure. On the trabecular surface, the three dimensional relationships are less clear, but the BMUs can be visualized as trenches of varying extent and depth.
Bone resorption begins with the removal of mineral and matrix together by a team of osteoclasts, excavating to a depth of about 50 µm (at a rate of about 5 µm/day) to form Howship's lacunae, which have scalloped border marking the foot prints of ruffled border of osteoclasts. The osteoclastic resorption extends longitudinally in cortical bone and across the trabecular plates in trabecular bone, at a rate of approximately 20 µm/day. After a brief period of quiescence, bone formation begins with the appearance of a team of mature cuboidal osteoblasts which lay down matrix that mineralizes after a lag period of about 5–10 days during which the collagen fibers and ground substance mature. The osteoblasts continue to move farther away with the continued matrix deposition, and the intervening unmineralized matrix is an osteoid seam which is normally about 10–15 µm in thickness. After completing the task of laying down new matrix to refill the excavated tunnel in cortical bone or a trench in trabecular bone, the osteoblasts undergo either apoptosis or become osteocytes buried within the mineralized bone.
At the junction of mineralized bone and osteoid is the zone of demarcation, a site where tetracycline and other bone seeking agents are incorporated, and thus, provide a useful method of studying dynamic histomorphometry.9 Apposition of matrix and bone mineral continues in parallel at about 1–1.5 µm/day, and the cells on the surface change from active to inactive osteoblasts. With this transition, there is also a considerable decline in matrix apposition, but a smaller decline in the rate of mineral apposition. Consequently, osteoid seam gradually gets thinner and eventually, disappears completely as the apposition of both matrix and mineral cease. Mineralization of the newly laid down matrix is most rapid in the initial phase, and approximately, 80% of the final mineral density is accomplished within a few days, referred to as primary mineralization. This is followed by a slow secondary mineralization that lasts about 3–6 months to reach a final mineral density of approximately 90%. Please note that bone is not fully mineralized to 100%, lest it will be dense but brittle, and the heterogeneity in the mean degree of mineralization confers unique biomechanical competence.4,6
At any point in time, about 15–20% of the bone surfaces are covered by a measurable osteoid seams representing sites of recent new bone formation, 70–75% of the surfaces are quiescent with flat lining cells, and the remaining surface is undergoing bone resorption. Every point on bone surface goes through successive stages quiescence, activation, resorption, reversal, and formation, and back to the quiescent state. It is estimated that bone remodeling occurs every 15 seconds at some place in the skeleton. In trabecular bone one such cycle occurs every 3 years and each cycle lasts about 3–4 months. It is important to appreciate that bone turnover at any point in time can be high, normal, or low, and each type of turnover can be associated with high, normal, or low bone density. This might sound paradoxical, but is similar to stock market or bank transactions; one can have a7 high-volume trading day (or deposits and withdrawals from a bank account) with falling prices (or bank balance) or a low volume trading with rising prices or any combination of the two. Also, somewhat analogous to financial transactions, it is important to appreciate the consequences of bone remodeling and turnover; small number of large resorption cavities, for instance, will have entirely different consequences than large number of small resorption cavities; the former causes irreversible structural damage because of the inability to recruit sufficient number of osteoblasts to refill the cavity within a finite time frame. Age, gender, and menopausal status each have significant influences on bone structure, remodeling, and osteoblast function.10–12
BONE HISTOLOGY AND HISTOMORPHOMETRY
It is essential to distinguish between these two seemingly confusing terms. Examination of bone tissue after decalcification is commonly used in clinical practice to determine the presence or absence of a nonmetabolic bone disease, and can be accomplished by standard methods without the need for specialized equipment for specimen preparation or staining procedures. Although used in this fashion for decades, decalcification of bone eliminates the opportunity to study mineralized and unmineralized bone fractions separately in a specimen, and does not provide detailed information about bone microarchitecture, remodeling, and bone cells. Dynamic bone histomorphometry, a method originally developed by late Dr Harold Frost, using double tetracycline labeling technique, revolutionized the study of bone biology leading to the “Frostian theory” of bone remodeling. Detailed histomorphometric study of bone is accomplished by obtaining a bone biopsy, usually from the anterior superior ilium, because of its easy access, following double tetracycline labeling.13,14 The biopsy specimen is then prepared with special techniques and stains to preserve the mineral and nonmineral components, and to keep the cellular details intact as they were at the time of the biopsy. The tetracycline fluorescence provides crucial information about the prevailing dynamic state of the bone at the time of biopsy, analogous to a time-lapse photography.14 Unfortunately, because of its invasive nature iliac bone biopsy and bone histomorphometry are used less often in evaluating patients with various metabolic bone diseases including osteoporosis. However, as a research tool, dynamic bone histomorphometry has contributed enormously to our current understanding of both normal bone status and of the pathogenesis of various forms of metabolic bone disorders, as well as defining the mechanism by which pharmacologic agents affect the bone. Detailed information on bone histomorphometric measurements and the nomenclature used has been published by the American Society for Bone and Mineral Research Task Force.9
SUMMARY OF SKELETAL HOMEOSTASIS
Maintenance of skeletal integrity, a prerequisite for optimal bone health, is a continual process accomplished by BMUs. Remodeling occurs in many tissues of the body including bone, otherwise, the bone like many nonbiologic materials (iron, steel, wood, etc.), would decay with time and crumble. Bone remodeling is a dynamic process occurring more frequently and efficiently than any other remodeling system in the body. It is estimated that every 15 seconds a piece of old bone is removed and replaced by new bone, ensuring skeletal youthfulness. The three types of cells, osteoclasts which initiate bone resorption to remove aged bone, osteoblasts which lay down new younger bone (both in quality and quantity), and osteocytes with their canalicular intercommunications, assure integrity and vitality of bone.8

FIG. 5: Conceptual (theoretical) optimal bone remodeling: Too high bone remodeling (shown on the right) creates too many holes in the bone (making it like a Swiss-cheese appearance) leading to trabecular perforation, loss of trabecular connectivity, and deteriorated microarchitecture that collectively increase susceptibility to fractures. On the other hand, too low remodeling (shown on the left) promotes crack propagation and microdamage accumulation, increases degree of mineralization (or hypermineralization), which collectively also increase susceptibility to fractures. Accordingly, bone remodeling needs to be at an optimal level just as the heart functions optimally at the midpoint of the well-known Frank–Starling curve (shown as black dot for both organs).
The bone remodeling rate in some ways is analogous to heart rate and function, which operates within a narrow range; tachycardia causes angina and bradycardia causes syncope, and the heart functions optimally at the “right” point on the Frank–Starling curve (Fig. 5). Similarly, too high remodeling results in architectural deterioration and too low remodeling promotes bone aging (not chronologic age of the individual) and microdamage accumulation (Fig. 5); both these extremes increase the fracture risk, but by different mechanisms. Fragility fractures due to microarchitectural deterioration occur after menopause as a result of too high remodeling, whereas atypical fractures occur after long-term severe suppression of bone turnover by pharmacologic agents.15 What is not known, however, is the “right” remodeling rate for optimal bone health. Bone cellular activity, although principally orchestrated by parathyroid hormone (PTH) and vitamin D, many systemic hormones, and locally produced cytokines and growth factors influence their number, life span, and function. This complex, but remarkably well-orchestrated system is essential not only to provide stature and locomotion, but also for the skeletal and mineral homeostatic functions of the skeleton.
Mineral Homeostasis
The human body is believed to consist of at least 24 elements, of which three divalent ions (calcium, phosphorus, and magnesium) are the most relevant for skeletal and mineral homeostasis. These three divalent ions, in addition to sodium and potassium, play crucial physiological roles in the regulation of electrical, mechanical, and biological functions of all tissues and organs in the body and in signal transductions. Consequently, mineral homeostatic system is highly regulated by the concerted actions of PTH, calcitonin, and 1,25-dihydroxyvitamin D, the active metabolite of vitamin D, on three major target organs—bone, kidney, and intestine. The physiological role of calcitonin in mineral homeostasis in humans remains largely unknown since, its deficiency after total thyroidectomy, or excess in medullary thyroid cancer and neuroendocrine tumors, rarely affects serum calcium, phosphate, or magnesium levels. However, calcitonin has important physiologic role in other mammals,16 and used pharmacologically in clinical practice.17 Although, largely independent from skeletal homeostasis, the mineral homeostatic system is subjected to perturbations both by physiologic changes (growth, aging, menarche, menopause, pregnancy, lactation, etc.), and pathologic conditions (fracture, humoral hypercalcemia9 of malignancy, tumor induced hypophosphatemia, direct tumor invasion of bone, renal failure, etc.). The system's steady-state is largely determined by the fluxes of minerals between blood [and extracellular fluid (ECF)] and bone, the blood/bone equilibrium,18 and by the intestinal absorption and renal excretion of divalent ions. Appreciation of these fundamental concepts underlying skeletal and mineral homeostatic systems is essential for understanding both the physiologic and pathologic changes in calcium, phosphorus, and to a lesser extent magnesium in the ECF.
Calcium: Distribution, Maintenance, and Measurement
Total body calcium content in humans is 1.0–1.2 g, of which 99% is in bone and the rest in ECF and soft tissues,19 and thus, depends entirely on the amount of bone. Despite this huge disproportionate distribution, calcium in both skeletal and extraskeletal compartments is tightly regulated by the respective homeostatic systems (vide supra). Any change in the external balance, therefore, depends entirely on the net movement of calcium in and out of bone; the blood/bone equilibrium,18,20 and on the intestinal absorption and renal excretion.21 Representative values for normal body content and distribution based on chemical analysis of cadavers are summarized in table 1, although the estimates by in vivo whole-body measurements in humans are approximately, 10–20% lower than the cadaveric analysis. Of the total bone calcium, only approximately 1% is in exchangeable pool with ECF.18 As evident, the small extraskeletal fraction (~1%) of calcium is distributed in muscle (~32%), ECF (~23%), and other tissues (~40%). Interestingly, red blood cells contain very tiny amounts of calcium (~0.0003 %), and so hemolysis rarely affects serum calcium measurement.
Of the total circulating calcium, about half is associated with various anions, mostly serum proteins, and about half is ionized.19 However, strictly speaking all calcium in the body must be ionized for its transport across membrane and cellular barriers, but the term ionized calcium is conventionally used to refer to the physiologically active free fraction in the circulation. A small fraction (~7%) of serum calcium is complexed by ion pairs with phosphate, bicarbonate, and citrate, but is freely diffusible across membranes; thus, the sum of complexed and ionized calcium constitutes the diffusible or ultrafilterable calcium.19 Thus, large load of citrate, as in massive blood transfusions22–25 or during acute dialysis26 measurement of total serum calcium can be misleading.
The steady state calcium balance is maintained by the net balance between the input from the intestinal absorption and output by the renal excretion with a small exchangeable pool between the blood and bone. Representative values for intake, absorption, excretion for calcium, phosphorus, and magnesium are shown in table 2. Intestinal absorption is both passive (paracellular, i.e., diffusion between cell junctions), and active (transcellular, i.e., across membranes); the latter is energy and vitamin D dependent.19 The bulk of the intestinal absorption occurs in the third and fourth parts of the duodenum and the proximal jejunum, and rapidly declines as a function of the length of the intestine with a small rise in the terminal ileum.
10
|
Both stomach and colon absorb minor quantities of calcium, but do not contribute significantly to serum calcium levels. However, pH in the stomach is a major determinant of ionization of calcium from its complex form in the food and supplements.27 Renal handling of calcium is also complex, and the total excretion by the kidney is determined by the net effects of the filter load (dependent on glomerular filtration rate) and renal tubular reabsorption (determined by sodium status and hormonal influences, PTH, ADH, vitamin D, etc.). Most of the filtered calcium in the kidney is reabsorbed in the proximal renal tubule (~70%), and the rest in the distal convoluted tubule (~20%) and collecting ducts (~10%).19 The proximal tubular reabsorption of calcium is sodium dependent and furosemide responsive, whereas the distal renal tubular reabsorption of calcium is sodium independent and thiazide responsive. This fundamental physiologic principle is the basis for the use of intravenous saline and furosemide, and avoidance of thiazide in hypercalcemic states,28,29 and the use of thiazides in idiopathic hypercalciuria and hypoparathyroidism.30 However, the “effect size” of both diuretics on serum calcium level is quite small,31 and “escapes” quickly (usually in days) once a new steady state is reached.
In all clinical chemistry laboratories throughout the world, serum total calcium is measured by spectrophotometers in multichannel auto-analyzers and ionized calcium is measured directly by ion selective electrodes.32 Due to the higher binding affinity of calcium to albumin (~80%), albumin adjusted serum calcium is preferred, although adjustment to either total protein or albumin is reasonable for most clinical situations.33,34 In conditions of excessive globulin synthesis (various gammopathies) or hypoalbuminemia (hospitalized patients, critically ill, elderly, etc.), measurement of ionized calcium is recommended.32,34 More importantly, the binding affinity of calcium to anions (protein, albumin, citrate, bicarbonate, etc.) depends heavily on the prevailing acid-base balance. Adjustment of measured calcium to total proteins or albumin, therefore, is not always consistent32,34 and is valid only within a narrow range (6.0–7.5 g/dL for total protein and 3.5–4.5 g/dL for albumin), as the slope of the relationship is less steep beyond these limits. Fortunately, the “effect size” of albumin adjustment to calcium is quite small (0.14–0.20 mg/dL), thus would not affect clinical decision making. Consequently, in most clinical situations albumin adjusted calcium is sufficient as the measurement of ionized calcium requires meticulous specimen handling, anaerobic condition, adjustment to prevailing pH, an ion selective electrode instrument, and is more expensive. In the author's institution, a computerized algorithm automatically adjusts serum calcium to albumin and is reported11 as part of the comprehensive metabolic profile. The practice has been in place since 1971 with necessary adjustments as the methods for measuring serum calcium and albumin have changed over the decades, and should really be adopted by all clinical chemistry laboratories.
Despite the challenges, serum calcium, however, measured or calculated, is relatively precise, and is tightly regulated and maintained within a narrow range with an interindividual standard deviation (SD) of only 0.32 mg/dL in healthy subjects,32,35,36 and with a much smaller intraindividual SD of ~0.10 mg/dL.37 There is a small diurnal variation in serum calcium level corresponding to the diurnal changes in PTH. Serum calcium is higher in children, increases after menopause (due to estrogen deficiency mediated increased bone resorption) and decreases during pregnancy (hemodilution). There is no age-related increase in serum calcium in men,38 but there is small, but significant difference in serum calcium levels between men and women,36 most likely related to the estrogen status. Due to the large reserve capacity of bone (about 99.5% of the total body calcium content) and the tightly controlled homeostatic system, serum calcium is maintained in the normal range with normal, high or low whole-body calcium balance, and/or with low, normal, or high bone turnover. Only when the blood/bone equilibrium is severely impaired, as in osteomalacia or excessive bone resorption of any kind is serum calcium low or high, respectively. In osteomalacia, accumulation of osteoid covering almost the bone entire surfaces effectively blocks the flux of calcium from bone to blood resulting in hypocalcemia.39 Conversely, in high resorptive states (humoral or tumor induced), the high flux of calcium from bone to blood exceeds the ability of the kidneys to excrete extra load resulting in hypercalcemia.40,41 Between these two extremes is the equilibrium hypercalcemia of primary hyperparathyroidism in which serum calcium remains relatively stable for extended periods;42 the interested reader is referred to an elegant exposition of the concept by Parfitt.18
Phosphate: Distribution and Maintenance
About approximately 85% of the total body phosphate is in bone and teeth, such that the total body content of phosphate, unlike that of total body calcium, is affected more by changes in soft tissue phosphate content than by changes in bone remodeling. However, extensive bone resorption as occurs in severe renal hyperparathyroidism can substantially increase both soft tissue and serum phosphate levels.43 Relative distribution of phosphate in various body compartments is summarized in table 1. Of the remaining 15% of the total body phosphate, 14% is intracellular and only 1% is extracellular. In the ECF, 70% of phosphate is in the organic (mostly phospholipids) and 30% in the inorganic form.44
Serum phosphate is also highly regulated, but not as tightly as serum calcium, primarily by the kidney mediated by the interactions of PTH and FGF-23, secreted almost exclusively by osteocytes.45 Unlike serum calcium, less than 10% of serum phosphate is bound to protein, but the specific proteins have not been identified, and a small fraction is complexed with calcium. Due to its critical role in various physiologic and energy functions, phosphate is mostly intracellular and needs to remain in the free form. Maintenance of serum phosphate is largely dependent on the renal handling and intestinal absorption and to a lesser extent by bone remodeling. For more detailed discussion (see chapter on Disorders of Phosphorus Metabolism).
Magnesium: Distribution and Maintenance
Among the cations in humans, magnesium is the second most abundant element intracellularly and fourth most abundant element in the body. Approximately, 60% of12 the total body magnesium is in the bone,19 consequently serum magnesium level is relatively less affected by changes in bone remodeling than serum calcium or phosphate levels; thus, the order of magnitude of change in serum levels of divalent ions influenced by bone remodeling is: calcium > phosphate > magnesium. Like serum calcium, serum magnesium is bound to proteins, mainly albumin, and the estimates of protein-bound magnesium vary between 20 and 40%. Relative distribution of magnesium in various body compartments is summarized in table 1. Magnesium is mainly intracellular, and the red cells contain proportionately higher magnesium than calcium.
Magnesium homeostasis is principally controlled by the kidney and handled differently along the length of the nephron. About 80% of the total serum magnesium is filtered through the kidney of which 5–15% is reabsorbed by the proximal renal tubule. This is distinct from sodium and calcium reabsorption in this segment of the nephron, where 70% of calcium and 60% of phosphate are reabsorbed. Proportionally greater amounts of magnesium (50–60%) are reabsorbed in the loop compared to sodium (20–25%) or calcium (30–35%); thus, the loop of Henle is the major determinant of magnesium reabsorption. Of the 10–15% of the filtered and delivered magnesium to the distal tubule, 70–80% is reabsorbed, leaving only about 3% of the filtered magnesium normally appearing in the urine.
Human magnesium deficiency is rare since any diet that sustains life is sufficient to provide the required daily intake except in extreme situations such as intestinal disease, resection, or bypass, alcoholic acute pancreatitis, renal tubular damage, and in the hospitalized patients.46 Similarly, hypermagnesemia is uncommon except in renal failure and hospitalized patients. The role of magnesium in skeletal and mineral homeostasis is not well understood as that of calcium and phosphate.47 However, hypomagnesemia, especially acutely, affects PTH secretion and synthesis.48
Several reports suggest an association between magnesium levels and various morbidities and mortality, but the effect size and the effect of replenishment have been inconsistent49–53 to the extent that magnesium is a required micronutrient in intestinal calcium absorption,54 a significant component of bone,19 and is involved in PTH secretion and synthesis, making it relevant in the evaluation of bone and mineral disorders in general,47 and hypoparathyroid states in particular.55–58 However, routine measurement of serum magnesium in clinical practice is not recommended, nor magnesium supplementation without confirmed magnesium deficiency.
SUMMARY OF MINERAL HOMEOSTASIS
Maintenance of calcium, phosphate, and magnesium is tightly regulated homeostatic system with concerted actions of various hormones acting on the three major target organs—bone, kidney, and intestine. The divalent cations circulate in the blood both in free and bound forms (mostly to albumin), but the proportion of binding differs. As various gammopathies interfere with their measurements by spectrophotometers, direct measurement by ion selective electrodes or reanalysis in protein free serum is recommended. Intraindividual variation of serum values are much narrower for all three divalent ions than interindividual variation. Serum calcium rises after menopause, but no effect of age in men. Serum phosphate is higher in children than in adults, most likely due to the effect of growth hormone on renal tubular reabsorption of phosphate. There is no age related change in serum magnesium. Consequently, age related reference ranges are necessary for phosphate, but not for serum calcium or magnesium levels. Phosphate and magnesium are predominantly intracellular, whereas calcium is predominantly extracellular. Bone remodeling plays a major role in modulating serum calcium, less so on13 serum phosphate and least on serum magnesium levels. Understanding these important physiological principles is essential to manage various pathological conditions altering divalent cations in the circulation.
SELECTED USEFUL WEBSITES
- American Society for Bone and Mineral Research (ASBMR): http://www.asbmr.org/education/bonecurriculum.aspx
- Bone Physiology and Pathology: http://courses.washington.edu/bonephys/ (A very useful website with photomicrographs and tables of most common metabolic bone diseases; a must visit site for all interested in metabolic bone diseases).
- Bone Pathology Index (a large collection of pathology slides).
REFERENCES
- Clarke B. Normal bone anatomy and physiology. Clin J Am Soc Nephrol. 2008;3(Suppl 3):S131–9.
- Baron R. Anatomy and ultrastructure of bone - histogenesis, growth and remodeling. In: De Groot LJ, Chrousos G, Dungan K, et al., editors. Endotext. South Dartmouth (MA): MDText.com, Inc.; 2000.
- Parfitt AM, Mathews CHE, Villanueva AR, et al. Relationships between surface, volume, and thickness of Iliac trabecular bone in aging and in Osteoporosis: Implications for the microanatomic and cellular mechanisms of bone loss. J Clin Invest. 1983;72:1396–409.
- Kular J, Tickner J, Chim SM, et al. An overview of the regulation of bone remodelling at the cellular level. Clinical Biochemistry. 2012;45(12):863–73.
- Valenti MT, Dalle Carbonare L, Mottes M. Osteogenic Differentiation in Healthy and Pathological Conditions. Int J Mol Sci. 2016;18(1).
- Parfitt AM. Quantum concept of bone remodeling and turnover: Implications for the Pathogenesis of Osteroporosis. Calcif Tissue Int. 1979;28:1–5.
- Dempster DW, Zhou H, Recker RR, et al. A longitudinal study of skeletal histomorphometry at 6 and 24 months across four bone envelopes in postmenopausal women with osteoporosis receiving teriparatide or zoledronic acid in the SHOTZ trial. J Bone Miner Res. 2016;31(7):1429–39.
- O'Brien CA, Nakashima T, Takayanagi H. Osteocyte control of osteoclastogenesis. Bone. 2013;54(2):258–63.
- Dempster DW, Compston JE, Drezner MK, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: A 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 2013;28(1):2–17.
- Han ZH, Palnitkar S, Rao DS, et al. Effect of ethnicity and age or menopause on the structure and geometry of iliac bone. J Bone Miner Res. 1996;11:1967–75.
- Han ZH, Palnitkar S, Rao DS, et al. Effects of ethnicity and age or menopause on the remodeling and turnover of iliac bone: implications for mechanisms of bone loss. J Bone Miner Res. 1997;12:498–508.
- Parfitt AM, Han ZH, Palnitkar S, et al. Effects of ethnicity and age or menopause on osteoblast function, bone mineralization, and osteoid accumulation in iliac bone. J Bone Miner Res. 1997;12:1864–73.
- Rao DS. Practical approach to bone biopsy. In: Recker R, editor. Bone histomorphometry: Techniques and interpretations. 1st ed. Boca Raton, FL: CRC Press; 1983. pp. 3–11.
- Vidal B, Pinto A, Galvao MJ, et al. Bone histomorphometry revisited. Acta Reumatol Port. 2012;37(4):294–300.
- Odvina CV, Zerwekh JE, Rao DS, et al. Severely suppressed bone turnover: A potential complication of alendronate therapy. J Clin Endocrinol Metab. 2005;90:1294–301.
- Rodriguez EM, Bach A, Devant M, et al. Is calcitonin an active hormone in the onset and prevention of hypocalcemia in dairy cattle? J Dairy Sci. 2016;99(4):3023–30.
- Bandeira L, Lewiecki EM, Bilezikian JP. Pharmacodynamics and pharmacokinetics of oral salmon calcitonin in the treatment of osteoporosis. Expert Opin Drug Metab Toxicol. 2016;12(6):681–9.
- Parfitt AM. Equilibrium and disequilibrium hypercalcemia new light on an old concept. Metab Bone Dis and Rel Res. 1979;1:279–93.
- Blaine J, Chonchol M, Levi M. Renal control of calcium, phosphate, and magnesium homeostasis. Clin J Am Soc Nephrol. 2015;10(7):1257–72.
- Felsenfeld AJ, Levine BS, Kleeman CR. Fuller Albright and our current understanding of calcium and phosphorus regulation and primary hyperparathyroidism. Nefrologia. 2011;31(3):346–57.
- Yu HY, O'Brien JJ, Magnani B. Conflicting calcium concentrations in the presence of low albumin after bone marrow transplantation. Clin Chem. 2010;56(11):1777–8.
- Das SS, Chaudhary R, Khetan D, et al. Calcium and magnesium levels during automated plateletpheresis in normal donors. Transfus Med. 2005;15(3):233–6.
- Giancarelli A, Birrer KL, Alban RF, et al. Hypocalcemia in trauma patients receiving massive transfusion. J Surg Res. 2016;202(1):182–7.
- Bicakci Z, Olcay L. Citrate metabolism and its complications in non-massive blood transfusions: Association with decompensated metabolic alkalosis+respiratory acidosis and serum electrolyte levels. Transfus Apher Sci. 2014;50(3):418–26.
- Feldkamp T, Weiler N, Marx M, et al. Critical deviations of ionized calcium measurements when using blood gas analyzers to monitor citrate dialysis. Clin Lab. 2016;62(10):2025–31.
- Keller J, Schinke T. The role of the gastrointestinal tract in calcium homeostasis and bone remodeling. Osteoporos Int. 2013;24(11):2737–48.
- Suki WN, Yium JJ, Von Minden M, et al. Acute treatment of hypercalcemia with furosemide. N Engl J Med. 1970;283(16):836–40.
- Parfitt AM. The interactions of thiazide diuretics with parathyroid hormone and vitamin D. Studies in patients with hypoparathyroidism. J Clin Invest. 1972;51(7):1879–88.
- Porter RH, Cox BG, Heaney D, et al. Treatment of hypoparathyroid patients with chlorthalidone. N Engl J Med. 1978;298(11):577–81.
- Wermers RA, Kearns AE, Jenkins GD, et al. Incidence and clinical spectrum of thiazide-associated hypercalcemia. Am J Med. 2007;120:911–5.
- Hughes D, Koerbin G, Potter JM, et al. Harmonising reference intervals for three calculated parameters used in clinical chemistry. Clin Biochem Rev. 2016;37(3):105–11.
- Scargill JJ, Guy JM. Validation of a locally derived serum calcium adjustment equation: Relationship with total 25(OH) vitamin D and PTH levels. J Clin Pathol. 2017;70(1):69–74.
- Mir AA, Goyal B, Datta SK, et al. Comparison between measured and calculated free calcium values at different serum albumin concentrations. J Lab Physicians. 2016;8(2):71–6.
- Rathod A, Bonny O, Guessous I, et al. Association of urinary calcium excretion with serum calcium and vitamin D levels. Clin J Am Soc Nephrol. 2015;10(3):452–62.
- Fijorek K, Puskulluoglu M, Tomaszewska D, et al. Serum potassium, sodium and calcium levels in healthy individuals - literature review and data analysis. Folia Med Cracov. 2014;54(1):53–70.
- Rao DS. Personal Observations. 2013.
- Tapernoux D, Diethelm M, Bucher C, et al. Influence of postmenopausal state, solar radiation, drugs and comorbidities on serum calcium and phosphate in 13,000 hospital admissions. Clin Nephrol. 2016;85(6):309–15.
- Basha B, Rao DS, Han ZH, et al. Osteomalacia due to vitamin D depletion in the US. Am J Med. 2000;108:296–300.
- Gottleib S, Rude RK, Sharp CF, et al. Humoral hypercalcemia of malignancy: A syndrome in search of a hormone. Am J Med. 1982;73(5):751–5.
- Stewart AF, Vignery A, Silverglate A, et al. Quantitative bone histomorphometry in humoral hypercalcemia of malignancy: Uncoupling of bone cell activity. J Clin Endocrinol Metab. 1982;55:219–27.
- Rao DS, Wilson RJ, Kleerekoper M, et al. Lack of biochemical progression or continuation of accelerated bone loss in mild asymptomatic primary hyperparathyroidism: evidence for biphasic disease course. J Clin Endocrinol Metab. 1988;67:1294–8.
- Moorthi RN, Moe SM. Recent advances in the noninvasive diagnosis of renal osteodystrophy. Kidney international. 2013;84(5):886–94.
- Osuka S, Razzaque MS. Can features of phosphate toxicity appear in normophosphatemia? J Bone Miner Metab. 2012;30(1):10–8.
- Jüppner H, Wolf M, Salusky IB. FGF-23: More than a regulator of renal phosphate handling? J Bone Miner Res. 2010;25(10):2091–7.
- Cheungpasitporn W, Thongprayoon C, Qian Q. Dysmagnesemia in hospitalized patients: Prevalence and prognostic importance. Mayo Clin Proc. 2015;90(8):1001–10.
- Rude RK, Singer FR, Gruber HE. Skeletal and hormonal effects of magnesium deficiency. J Am Coll Nutr. 2009;28(2):131–41.
- Hoorn EJ, Zietse R. Disorders of calcium and magnesium balance: A physiology-based approach. Pediatr Nephrol. 2013;28(8):1195–206.
- Yu L, Li H, Wang SX. Serum magnesium and mortality in maintenance hemodialysis patients. Blood Purif. 2017;43(1-3):31–6.
- Guerrero-Romero F, Jaquez-Chairez FO, Rodriguez-Moran M. Magnesium in metabolic syndrome: A review based on randomized, double-blind clinical trials. Magnes Res. 2016;29(4):146–53.
- Naksuk N, Hu T, Krittanawong C, et al. Association of serum magnesium on mortality in patients admitted to the intensive cardiac care unit. Am J Med. 2017;130(2):229.e5-e13.
- Osborn KE, Shytle RD, Frontera AT, et al. Addressing potential role of magnesium dyshomeostasis to improve treatment efficacy for epilepsy: A reexamination of the literature. J Clin Pharmacol. 2016;56(3):260–5.
- Heaton FW, Hodgkinson A, Rose GA. Observations on the relation between calcium and magnesium metabolism in man. Clin Sci. 1964;27:31–40.
- Habener JF, Potts JT, Jr. Relative effectiveness of magnesium and calcium on the secretion and biosynthesis of parathyroid hormone in vitro. Endocrinology. 1976;98(1):197–202.
- Michelis MF, Bragdon RW, Fusco RD, et al. Parathyroid hormone responsiveness in hypoparathyroidism with hypomagnesemia. Am J Med Sci. 1975;270(3):412–8.
- Wiegmann T, Kaye M. Hypomagnesemic hypocalcemia. Early serum calcium and late parathyroid hormone increase with magnesium therapy. Arch Intern Med. 1977;137(7):953–5.
- Rude RK, Oldham SB, Singer FR. Functional hypoparathyroidism and parathyroid hormone end-organ resistance in human magnesium deficiency. Clin Endocrinol. 1976;5(3):209–24.