

ORIGIN OF DOSAGE FORMS
The origins of the earliest dosage forms are lost in the mists of history. We can safely assume that primitive man took parts of plants including leaves, stems, roots and berries internally for a range of symptoms. A variety of plant and animal products would undoubtedly also have been applied externally to aid the healing of wounds. The vapors of volatile herbs would have been inhaled, and later combinations would have been used, no doubt incorporated into a selection of fats, oils and honey. As time went on they would have been processed by boiling, grinding or dissolving.
Early civilizations used an extensive range of dosage forms, for the application of medicines to every surface and orifice. The Ancient Greeks and Romans used a number of dosage forms, many of which we would recognize today: They included ointments, oils, powders, pills, pessaries, gargles and eye lotions. Others had different names to those used today; enemas were known as clysters or glisters, and were liquids that were injected into the rectum using a horn as a funnel, or an animal bladder attached to a greased tube inserted into the anus.
The Arabs introduced a number of sweet preparations based on sugar, including syrups, conserves, confections, electuaries and juleps. Syrups were made by boiling an extract with sugar; conserves were flowers, herbs, roots or fruits preserved in sugar; confections and electuaries both involved mixing dried and powdered ingredients with syrup or honey; and juleps were clear, sweet liquids. The Arabs also used flavors such as rose water, orange and lemon peel.
Following the departure of the Romans from Britain in the early fifth century, Anglo-Saxon practitioners used a form of medicine known as leeches. Their preparations included ointments, poultices, plasters, fomentations, internal medicines sweetened with honey, herbs mixed 2with water, ale or wine, inhalations of vapors, and fumigation using hot herbal decoctions or burning seeds.
In the fifteenth century, the introduction of printing meant that books such as pharmacopoeias became more widely available. These contained formulas for frequently-prescribed medicines and also methods for making them. As well as herb simples, containing a single remedy, they included many medicines with a large number of ingredients.
Medicines for internal use ranged in consistency from watery liquids to thick liquids, such as syrups and emulsions, to semi-solid products such as lozenges and pastilles, to solid dose forms such as pills and tablets. Early liquid medicines included spirits and distilled waters, which were made by macerating drugs with spirit of wine and then collecting the liquid produced by distillation. Mixtures of oils were distilled to produce balsams, whilst quintessences were produced by distilling an essential oil in pure spirit of wine and concentrating the result by repeated distillation. Tinctures were made by soaking the ingredient in spirit and then straining the result.
Elixirs were similar to tinctures, but stronger and thicker. Powders were preparations made from ingredients that were mixed together and powdered in a mortar and pestle. They were either a simple (with one ingredient) or compound. As they often had an unpleasant taste, pharmacists would insert them between layers of rice paper, becoming a wafer. An improvement on wafers was cachets, consisting of powder sealed between shaped rice paper plates.
Pills were a useful dosage form for medicines that had an unpleasant taste. Solid ingredients were powdered and then made into a stiff mass, which was then formed into roughly spherical pills. A variety of equipment was developed to facilitate this, and pill-making became an essential part of the pharmacist's art.
Mass production enabled the development of products with reliable content and characteristics. Capsules and tablets became the dominant forms. Products were also developed to be dissolved in the mouth rather than to be swallowed. These included lozenges and pastilles.
Amongst the preparations applied externally were ointments, creams and plasters. Ointments were made using several types of base: greasy ones made from fats, waxes or oils; emulsifying bases that could be emulsified by water, oil-in-water or water-in-oil emulsion bases; and water-soluble bases. Today they have largely been replaced by greasy or non-greasy creams.
3Lotions were liquid external preparations designed to be applied without friction, whereas liniments were designed to be rubbed into the skin. Pastes were topical preparations in a base of starch, glycerin, glycogelatin or paraffin.
Plasters (not to be confused with sticking plasters) were usually spread on a fabric such as leather or calico using a hot plaster iron. Shapes for use on the breast, back, chest or shoulder were made.
As well as application to the skin, products were developed for insertion into body cavities. These included suppositories, pessaries and bougies. Suppositories are solid preparations made by mixing the active ingredient with molten theobroma oil, glycerin or other material, pouring into moulds and allowing to set. They are normally bullet-shaped to allow easy insertion into the rectum. Pessaries are for vaginal insertion, and bougies were medicated pencils for urethral, aural or nasal use.
Enemas are liquid preparations for insertion into the rectum, and are mainly used to relieve severe constipation. Originally powerful clyster syringes were used, but these were replaced by a funnel and rubber tube and more recently by microenemas.
Lamellae were medicated discs intended for application to the eye; they were made from glycerin, gelatin and water in which the active ingredient was dissolved. Inhalations were volatile substances that could be inhaled from a handkerchief or inhaler. Others were dispersed in hot water and inhaled from a container such as a Nelson's inhaler. Vitrellae were capsules made of thin glass that could be crushed and the contents inhaled. Finally, injections were sterile products that could be injected by a number of routes including intramuscular, intravenous and subcutaneous.
TYPES OF COMMONLY USED DOSAGE FORMS
Dosage form is a transformation of a pure chemical compound by processing into a predetermined form by admixing drug component with different kinds of inert non-drug components. Hence dosage forms are the means by which drug molecules are delivered to sites of action within the body. Dosage form can be classified as-liquid, solid and semisolid.
A dosage form is the physical form of a dose of medication, such as a capsule or injection. The route of administration is dependent on the dosage form of a given drug. Various dosage forms may exist for 4the same compound, since different medical conditions may warrant different routes of administration. For example, persistent vomiting may make it difficult to use an oral dosage form; in this case, it may be advisable to use either an injection or a suppository. Also, specific dosage forms may be warranted for certain medications, since there may be problems with stability, e.g. insulin cannot be given orally since it is digested by the gut.
To achieve an optimum response from any dosage form, a drug should be delivered to its site of action at a rate and concentration that both minimize its side effects and maximize its therapeutic effects. The development of safe and effective drug dosage forms and delivery systems requires a thorough understanding of physicochemical principles that allow a drug to be formulated into a pharmaceutical dosage form. Development of the appropriate dosage form or delivery system depends on the:
- Physicochemical properties of the drug, such as solubility, oil-to-water partition coefficient (Ko/w), pKa value, and molecular weight
- Dose of the drug
- Route of administration
- Type of dosage form/drug delivery system desired
- Pathologic condition to be treated
- Desired therapeutic effect
- Drug release from the delivery system
- Bioavailability of the drug at the absorption site
- Pharmacokinetics and pharmacodynamics of the drug.
Solutions, Syrups, and Elixirs
Solutions are homogeneous mixtures of one or more solutes dispersed in a dissolving medium (solvent). Aqueous solutions containing a sugar or sugar substitute with or without added flavoring agents and drugs are classified as syrups. Sweetened hydroalcoholic (combinations of water and ethanol) solutions are termed elixirs. Hydroalcoholic solutions of aromatic materials are termed spirits. Solutions intended for oral administration usually contain flavorants and colorants to make the medication more attractive and palatable to the patient. They may contain stabilizers to maintain the physicochemical stability of the drug and preservatives to prevent the growth of microorganisms in the solution. A drug dissolved in an aqueous solution is in the most bioavailable form. Since the drug is already in solution, no dissolution 5step is necessary before systemic absorption occurs. Solutions that are prepared to be sterile, pyrogen-free, and intended for parenteral administration are classified as injectables.
Some drugs, particularly certain antibiotics, have insufficient stability in aqueous solution to withstand long shelf-lives. These drugs, formulated as dry powder or granule dosage forms, are reconstituted with purified water immediately before dispensing to the patient. The dry powder mixture contains all of the formulation components (i.e. drug, flavorant, colorant, buffers, and others, except for the solvent). Examples of dry powder mixtures intended for reconstitution to make oral solutions include cloxacillin sodium, nafcillin sodium, oxacillin sodium, penicillin V potassium, and potassium chloride.
Sucrose is the sugar most frequently employed in syrups; in special circumstances it may be replaced in whole or in part by other sugars (e.g. dextrose) or nonsugars (e.g. sorbitol, glycerin, and propylene glycol). Most syrup consists of between 60 and 80% sucrose. Sucrose not only provides sweetness and viscosity to the solution; it renders the solution inherently stable (unlike dilute sucrose solutions, which are unstable).
Elixirs are usually less sweet and less viscous than syrups. Since elixirs contain a lower proportion of sugar, they are consequently less effective than syrups in masking the taste of drugs. In contrast to aqueous syrups, elixirs are better able to maintain both water-soluble and alcohol-soluble components in solution due to their hydroalcoholic properties. These stable characteristics often make elixirs preferable to syrups.
All elixirs contain flavoring and coloring agents to enhance their palatability and appearance. Elixirs containing over 10–12% alcohol are usually self-preserving and do not require the addition of antimicrobial agents for preservation. Alcohols precipitate tragacanth, acacia, agar, and inorganic salts from aqueous solutions; therefore such substances should either be absent from the aqueous phase or present in such low concentrations so as not to promote precipitation on standing. Examples of some commonly used elixirs include dexamethasone elixir USP, pentobarbital elixir USP, diphenhydramine hydrochloride elixir, and digoxin elixir.
Tablets
Depending on the physicochemical properties of the drug, site and extent of drug absorption in the gastrointestinal (GI) tract, stability to heat or moisture, biocompatibility with other ingredients, solubility, and dose, the following types of tablets are commonly formulated:
- Effervescent tablets that need to be dissolved in water prior to administration.
- Chewable tablets are used when a faster rate of dissolution and/or buccal absorption is desired. Chewable tablets consist of a mild effervescent drug complex dispersed throughout a gum base. The drug is released from the dosage form by physical disruption associated with chewing, chemical disruption caused by the interaction with the fluids in the oral cavity, and the presence of effervescent material. For example, antacid tablets should be chewed to obtain quick indigestion relief.
- Buccal and sublingual tablets dissolve slowly in the mouth, cheek pouch (buccal), or under the tongue (sublingual). Buccal or sublingual absorption is often desirable for drugs subject to extensive hepatic metabolism, often referred to as the first-pass effect. Examples are isoprenaline sulfate (bronchodilator), glyceryl trinitrate (vasodilator), nitroglycerin, and testosterone tablets. These tablets do not contain a disintegrant and are compressed lightly to produce a fairly soft tablet.
- Controlled-release tablets are used to improve patient compliance and to reduce side effects. Some water-soluble drugs are formulated as sustained release tablets, so that their release and dissolution is controlled over a long period. A hydrophobic matrix composed of carnauba wax and partially hydrogenated cottonseed oil were used to prepare sustained-release tablets of a highly water-soluble drug, ABT-089, a cholinergic channel modulator for the treatment of cognitive disorders. Theo-Dur is a controlled-release tablet of theophylline and consists of two components: a matrix of compressed theophylline crystals and coated theophylline granules embedded in the matrix. In contact with fluid, theophylline diffuses slowly through the wall of the free granules, which dissolves with time. After oral administration of Theo-Dur 300 mg tablets to human subjects, serum theophylline concentrations over 1 mg/mL were maintained over 24 hours. To provide a zero-order release of ibuprofen, core-in-cup tablets were developed by compressing the mixture of ethyl cellulose and carnauba wax, followed by compression with core tablets containing ibuprofen. Drugs with unpleasant flavors that irritate the stomach walls are formulated inside the tablets so as to be bound to ion-exchange 7resins and coated with a water-insoluble polymer membrane made of ethylcellulose or a similar substance. These drugs include codeine, dextromethorphan, acetaminophen, ephedrine, and chlorpheniramine. Aspirin has been shown to produce less gastric bleeding when formulated as a sustained-release formulation than conventional tablets. The combination of high- and low-viscosity grades of hydroxypropyl methylcellulose (HPMC) was used as the matrix base to prepare diclofenac sodium and zileuton sustained-release tablets. A ternary polymeric matrix system composed of protein, HPMC, and highly water-soluble drugs such as diltiazem hydrochloride was developed by the direct compression method. Xanthan gum was used for a hydrophilic matrix for sustained release ibuprofen tablets. Sustained-release tablets can also be prepared by formulating inert polymers like polyvinyl chloride, polyvinyl acetate, and methyl methacrylate. These polymers protect the tablet from disintegration and also reduce the dissolution rate of the drug inside the tablet.
- Coated tablets: There are several types of coated tablets: film coated, sugar coated, gelatin coated (gel caps), or enteric-coated tablets. Enteric coatings are resistant to gastric juices, but readily dissolve in the small intestine. These enteric coatings can protect drugs against decomposition in the acid environment of the stomach. Commonly used polymers for enteric coating are acid-impermeable polymers, such as cellulose acetate trimellitate (CAT), hydroxypropyl methylcellulose phthalate (HPMCP), polyvinyl acetate phthalate (PVAP), cellulose acetate phthalate (CAP) and Eudragit. Aspirin has been shown to produce less gastric bleeding when formulated as enteric-coated sustained-release tablets than conventional aspirin preparations. Film-coated tablets are compressed tablets that are coated with a thin layer of a water insoluble or water soluble polymer, such as HPMC, ethylcellulose, povidone or polyethylene glycol. AbacavirTM is a capsule-shaped film-coated tablet containing a nucleoside reverse transcriptase inhibitor, which is a potent antiviral agent for the treatment of HIV infection.
Capsules
Capsules are solid dosage forms in which the drug substance is enclosed in either a hard or soft, water-soluble container or shell of gelatin. Coating of capsule shell or drug particles within the capsule can affect 8bioavailability. There are two types of capsules: Hard and soft capsules; however, hard gelatin capsules are more versatile for controlled drug delivery.
- Hard gelatin capsule consists of two pieces, a cap and a body, that fit one inside the other. They are produced empty and are then filled in a separate operation. Hard gelatin capsules are usually filled with powders, granules, or pellets containing the drug. After ingestion, the gelatin shell softens, swells, and begins to dissolve in the gastrointestinal tract. Encapsulated drugs are released rapidly and dispersed easily, leading to high bioavailability. Capsules are supplied in a variety of sizes, and high-speed filling machinery capable of filling ~1500 capsules per minute is available. The hard gelatin empty capsules are numbered from 000, the largest size, to 5, which is the smallest. The approximate filling capacity of capsules ranges from 6000 to 30 mg, depending on the types and bulk densities of powdered drug materials.
- Soft gelatin capsules are prepared from plasticized gelatin by a rotary die process in which they are formed, filled, and sealed in a single operation. Soft gelatin capsules may contain a non-aqueous solution, a powder, or a drug suspension, none of which solubilize the gelatin shell. In contrast to hard gelatin capsules, soft gelatin capsules contain ~30% glycerol as a plasticizer in addition to gelatin and water. The moisture uptake of soft gelatin capsules plasticized with glycerol is considerably higher than that of hard gelatin capsules. Therefore, oxygen-sensitive drugs should not be inserted into soft gelatin capsules; nor should emulsions, since they are unstable and crack the shell of the capsule when the water is lost in the manufacturing process. Extreme acidic and basic pH must also be avoided, since a pH below 2.5 hydrolyzes gelatin, while a pH above 9 has a tanning effect on the gelatin. Insoluble drugs should be dispersed with an agent such as beeswax, paraffin, or ethylcellulose. Surfactants are also often added to promote wetting of the ingredients. Drugs that are commercially prepared in soft capsules include declomycin, chlorotrianisene, digoxin, vitamin A, vitamin E, and chloral hydrate.
Emulsions
An emulsion is a thermodynamically unstable system consisting of at least two immiscible liquid phases, one of which is dispersed as globules (dispersed phase) in the other liquid phase (continuous 9phase), stabilized by the presence of an emulsifying agent. Emulsified systems range from lotions of relatively low viscosity, to creams, which are semisolid in nature.
Two immiscible liquids in emulsions often fail to remain mixed due to the greater cohesive force between the molecules of each separate liquid than the adhesive force between the two liquids. This leads to phase separation, which is the state of minimum surface free energy. When one liquid is broken into small particles, the interfacial area of the globules constitutes a surface that is enormous compared with the surface area of the original liquid. The adsorption of a surfactant or other emulsifying agent at the globule interface lowers the oil-to-water or water-to-oil interfacial tension. In addition, the process of emulsification is made easier and the drug's stability may be enhanced.
- Oil-in-water (o/w) emulsion: When the oil phase is dispersed as globules throughout an aqueous continuous phase, the system is referred to as an oil-in-water (o/w) emulsion.
- Water-in-oil (w/o) emulsion: When the oil phase serves as the continuous phase, the emulsion is termed water-in-oil (w/o) emulsion.
- Multiple (w/o/w or o/w/o) emulsions: These are emulsions whose dispersed phase contains droplets of another phase. Multiple emulsions are of interest as delayed-action drug delivery systems.
- Microemulsions: These consist of homogeneous transparent systems of low viscosity which contain a high percentage of both oil and water and high concentrations of emulsifier mixture. Microemulsions form spontaneously when the components are mixed in the appropriate ratios and are thermodynamically stable.
Externally applied emulsions may be o/w or w/o. The o/w emulsions employ these types of emulsifiers: Sodium lauryl sulfate, triethanolamine stearate, sodium oleate, and glyceryl monostearate. The w/o emulsions are used mainly for external applications and may contain one or several of these emulsifiers: Calcium palmitate, sorbitan esters (Spans), cholesterol, and wool fats.
Suspensions
Suspensions are dispersions of finely divided solid particles of a drug in a liquid medium in which the drug is not readily soluble. Suspending agents are often hydrophilic colloids (e.g. cellulose derivatives, acacia, or xanthan gum) added to suspensions to increase viscosity, inhibit agglomeration, and decrease sedimentation. Highly viscous 10suspensions may prolong gastric emptying time, slow drug dissolution, and decrease the absorption rate. A suspension that is thixotropic as well as pseudoplastic should prove to be useful since it forms a gel on standing and becomes fluid when disturbed.
Suspended material should not settle rapidly. The particles that do settle on the bottom should not form a hard cake, but should be readily redispersed into a uniform mixture when shaken. Aggregation can be prevented if the particles have a similar electrical charge.
The large surface area of the particles is associated with a surface free energy that makes the system thermodynamically unstable. This makes particles highly energetic and tends to regroup, resulting in the decrease in total surface area and surface free energy. The particles in a liquid suspension, therefore, tend to flocculate. Flocculation is the formation of light, fluffy conglomerates held together by weak van der Waals forces. Aggregation occurs in a compact cake situation (growth and fusing together of crystals in the precipitates to form a solid aggregate). Flocculating agents can prevent caking, whereas deflocculating agents increase the tendency to cake. Surfactants can reduce interfacial tension, but it cannot be made equal to zero, so suspensions of insoluble particles tend to have a positive finite interfacial tension, and particles tend to flocculate (Table 1.1).
Forces at the surface of a particle affect the degree of flocculation and agglomeration in a suspension. Forces of attraction are of the London van der Waals type, whereas the repulsive forces arise from the interaction of the electric double layers surrounding each particle.
When the repulsion energy is high, collision of the particles is opposed; the 11system remains deflocculated and when sedimentation is complete, the particles form a close-packed arrangement with the smaller particles filling the void between the larger ones. Those particles lowest in the sediment are gradually pressed together by the weight of the ones above; the energy barrier is thus overcome, allowing the particles to come into close contact with each other. To resuspend and redisperse these particles, it is necessary to overcome the high energy barrier. Since this is not easily achieved by agitation, the particles tend to remain strongly attracted to each other and form a hard cake. When the particles are flocculated, the energy barrier is still too large to be surmounted, and so the approaching particles in the second energy minimum, which is at a distance of separation of perhaps 1000 to 2000 Å, is sufficient to form the loosely structural flocs.
Flocs tend to fall together, producing a distinct boundary between the sediment and the supernatant liquid. The liquid above the sediment is clear because even the small particles present in the system are associated with flocs. In contrast to this are deflocculated systems with variable particle sizes; the large particles here settle more rapidly than the smaller particles, and no clear boundary is formed. The supernatant remains turbid for a longer period of time.
There are two ways of formulating physically stable suspensions:
- The use of a structured vehicle to maintain deflocculated particles in suspension. However, the major disadvantage of deflocculated systems is that when the particles eventually settle, they form a compact cake.
- Production of flocs, which may settle rapidly, but are easily resuspended with a minimum of agitation. Optimum physical stability is obtained when the suspension is formulated with flocculated particles in a structured vehicle of hydrophilic colloid type.
Ointments
Ointments are semisolid preparations intended for external application. Ointments are typically used as:
- Emollients to make the skin more pliable
- Protective barriers to prevent harmful substances from coming in contact with the skin
- Vehicles in which to incorporate medication.
Ointment bases are classified into four general groups: Hydrocarbon bases, absorption bases, water-removable bases, and water-soluble bases.
12Drugs may be incorporated into an ointment base by levigation and fusion. Normally, drug substances are in fine powered forms before being dispersed in the vehicle. Levigation of powders into a small portion of base is facilitated by the use of a melted base or a small quantity of compatible levigation aid, such as mineral oil or glycerin. Water-soluble salts are incorporated by dissolving them in a small volume of water and incorporating the aqueous solution into a compatible base. Fusion method is used when the base contains solids that have higher melting points (e.g. waxes, cetyl alcohol, or glyceryl monostearate).
Creams
A cream is a topical preparation usually for application to the skin. Creams for application to mucous membranes such as those of the rectum or vagina are also used. Creams may be considered pharmaceutical products as even cosmetic creams are based on techniques developed by pharmacy and unmedicated creams are highly used in a variety of skin conditions (dermatoses).
Creams are semi-solid emulsions that are mixtures of oil and water. They are divided into two types: oil-in-water (o/w) creams which are composed of small droplets of oil dispersed in a continuous aqueous phase, and water-in-oil (w/o) creams which are composed of small droplets of water dispersed in a continuous oily phase. Oil-in-water creams are more comfortable and cosmetically acceptable as they are less greasy and more easily washed off using water. Water-in-oil creams are more difficult to handle but many drugs which are incorporated into creams are hydrophobic and will be released more readily from a water-in-oil cream than an oil-in-water cream. Water-in-oil creams are also more moisturizing as they provide an oily barrier which reduces water loss from the stratum corneum, the outmost layer of the skin. The uses of creams include:
- The provision of a barrier to protect the skin
- To aid in the retention of moisture (especially water-in-oil creams)
- Cleansing action
- Emollient effects
- As a vehicle for drug substances such as local anaesthetics, anti-inflammatory (NSAIDs or corticosteroids), hormones, antibiotics, antifungal or counter-irritants.
Gels
13A gel is a solid, jelly-like material that can have properties ranging from soft and weak to hard and tough. Gels are defined as a substantially dilute cross-linked system, which exhibits no flow when in the steady-state. By weight, gels are mostly liquid, yet they behave like solids due to a three-dimensional cross-linked network within the liquid. It is the crosslink's within the fluid that gives a gel its structure (hardness) and contribute to stickiness (tack).
- Hydrogel (also called Aquagel) is a network of polymer chains that are water-insoluble, sometimes found as a colloidal gel in which water is the dispersion medium. Hydrogels are superabsorbent (they can contain over 99% water) natural or synthetic polymers. Hydrogels also possess a degree of flexibility very similar to natural tissue, due to their significant water content.
- Organogel is a non-crystalline, non-glassy thermoreversible (thermoplastic) solid material composed of a liquid organic phase entrapped in a three-dimensionally cross-linked network. The liquid can be, e.g. an organic solvent, a mineral oil or a vegetable oil. The solubility and particle dimensions of the structurant are important characteristics for the elastic properties and firmness of the organogel. Often, these systems are based on self-assembly of the structurant molecules.
Gels and jellies serves as vehicles for topically applied drugs as emollients or as protective or occlusive dressings. They are also applied to mucous membrane such as rectal, vaginal.
Ophthalmic Preparations
Drugs are administered to the eye for local effects such as miosis, mydriasis, and anesthesia, or to reduce intraocular pressure in treating glaucoma. The ophthalmic formulation delivers the drug on the eye, into the eye, or onto the conjunctiva. Transcorneal transport (i.e. drug penetration into the eye) is not an effective process. It is estimated that only one-tenth of a dose penetrates into the eye.
Ophthalmic formulations used include aqueous solutions, aqueous suspensions, ointments, and inserts. Every ophthalmic product must be sterile in its final container to prevent microbial contamination of the eye. Preservatives are added to the formulation to maintain sterility once the container has been opened. Ophthalmic formulations also require 14that the pH, buffer capacity, viscosity, and tonicity of the formulation be carefully controlled.
Ophthalmic solutions such as eye drops must be free from foreign particles, and specially prepared for instillation in the eye. Most ophthalmic solutions are dispensed in eye dropper bottles. Patients should be shown how to properly instill the drops in their eyes, and every effort should be made to emphasize the need for instilling only one drop per administration, not two or three. When more than one drop is to be administered, wait at least five minutes between administrations. Immediately after instilling a drop on the eye, place pressure on the lachrymal sac for one or two minutes. This will reduce the rate of drug loss through this pathway.
Ophthalmic suspensions are aqueous formulations that contain solid particles. The particle size must be kept to a minimum to prevent irritation of the eye. It has been recommended that particles be less than 10 microns in size to minimize irritation to the eye. The micronized form of the drug can be used to meet this requirement. There is a tendency of the solid undissolved particles to adhere to the conjunctiva. As drug is absorbed, these solid particles will dissolve to replenish the absorbed drug. This reservoir effect increases the contact time and duration of action of a suspension compared to a solution.
In an effort to maintain longer contact between the drug and ocular tissue, ointments and inserts have been used. Ophthalmic ointments tend to keep the drug in contact with the eye longer than suspensions. Most ophthalmic ointment bases are a mixture of mineral oil and white petrolatum and have a melting point close to body temperature. Sometimes anhydrous lanolin is used to take up an ingredient that was dissolved in a small amount of water to affect dissolution. The aqueous solution is incorporated into the lanolin and then the lanolin is mixed with the remaining ointment base ingredients.
Parenteral Preparations (Injectable Preparations)
These are sterile dosage forms containing one or more medicaments and designed for parenteral administration. Parenteral preparations or injectables are the sterile solutions or suspensions of drugs in aqueous vehicle or oily vehicles meant for introduction into the body by means of an injection under or through one or more layers of skin or mucous membrane. They must be sterile and free from all types of living microorganisms and microbial products such as toxins, pyrogens, etc. and should be free from particles like fibers, dust, etc. They should be 15isotonic with body fluids. They should be introduced through the same route for which they are intended.
- Injections are sterile solutions, suspensions or emulsions in a suitable aqueous or non-aqueous vehicle and are usually classified according to their route of administration (Table 1.2).
- Powders for injections are sterile solid substances to be dissolved or suspended by adding a prescribed volume of the appropriate sterile fluid. The solution or suspension is usually prepared immediately prior to use to avoid deterioration of the product on storage.
- Intravenous infusions are sterile aqueous solutions or emulsions, free from pyrogens and usually made isotonic with blood. They do not contain added antimicrobial preservatives or buffering agents and are designed for intravenous administration in volumes usually greater than 10–15 mL.
Parenteral preparations can be classified as:
- Sterile solutions: They are commonly called injections. Such parenteral preparations are supplied in single dose container or multiple dose containers, e.g. atropine sulfate injection, dextrose injection.
Table 1.2 A brief list of route of administration of parenteral preparations RouteMeaningExamplesIntravenousInto a veinMany drugs, total parenteral nutritionIntra-arterialInto an arteryVasodilator drugs in the treatment of vasospasm and thrombolytic drugs for treatment of embolismIntramuscularInto a muscleMany vaccines, antibiotics, and long-term psychoactive agentsIntracardiacInto the heartAdrenaline during cardiopulmonary resuscitationSubcutaneousUnder the skinInsulinIntradermalInto the skin itselfUsed for skin testing some allergensIntraosseous infusionInto the bone marrowOccasionally used for drugs and fluids in emergency medicine and pediatrics when intravenous access is difficultIntrathecalInto the spinal canalCommonly used for spinal anesthesia and chemotherapyIntracisternalBetween first and second cervical vertebraeUsed to withdraw CSF for diagnostic purposesIntraperitonealInfusion or injection into the peritoneumPeritoneal dialysisIntracerebralInto cerebrum-- - 16Sterile suspensions: These are sterile suspension of drugs in a suitable solvent, which are administered by intramuscular route. The solid content of parenteral suspensions usually ranges between 0.5 and 5% but may go as high as 30% in some antibiotic preparations, e.g. sterile hydrocortisone acetate suspension, sterile chloramphenicol suspension.
- Sterile emulsions: These are sterile emulsions of drugs and the internal phase is in the form of uniform oil droplets in the size of 1 to 5 microns, e.g. emulsion of vitamin K1, intravenous nutrient emulsions.
- Dry powder formulations: These are lyophilized or freeze-dried powders that must be reconstituted with some suitable solvent to make a liquid formulation before being withdrawn from the vial. Some drugs are not stable in liquid form and so these drugs are put into the powder form and reconstituted just prior to use. There are several solvents that might be used to reconstitute the dry powders; the appropriate solvent is indicated in the product information insert. The most common solvents are sterile water for injection, bacteriostatic water for injection, sodium chloride injection, and Ringer's injection.
- Long-acting formulations: These are designed to provide slow, constant and sustained release of a drug over a prolonged period of time. Depot formulations have been developed to achieve the desired goal.
- Transfusion fluids: These are parenteral preparations that are administered by intravenous route and are used for nutrition and to maintain the electrolyte balance, e.g. sodium chloride injection, dextrose injection.
Aerosol Products
Aerosols are pressurized dosage forms designed to deliver drugs with the aid of a liquefied or propelled gas (propellant). Aerosol products consist of a pressurizable container, a valve that allows the pressurized product to be expelled from the container when the actuator is pressed, and a dip tube that conveys the formulation from the bottom of the container to the valve assembly. Inhalation devices broadly fall into three categories: pressurized metered-dose inhalers (MDIs), nebulizers, 17and dry-powder inhalers (DPIs). The most commonly used inhalers on the market are MDIs. They contain active ingredient as a solution or as a suspension of fine particles in a liquefied propellant held under high pressure. MDIs use special metering valves to regulate the amount of formulation that is dispensed with each dose. Nebulizers do not require propellants and can generate large quantities of small droplets capable of penetrating into the lung. Sustained release of drugs, such as bronchodilators and corticosteroids for the treatment of asthma and chronic obstructive pulmonary diseases, involves encapsulation of the drugs in slowly degrading particles that can be inhaled. For accumulation in the alveolar zone of the lungs, which has very large surface area, inhaled liquid or dry-powder aerosols should have particle sizes in the range of 1–5 μm. Inhaled drugs play a very prominent role in the treatment of asthma, because this route has significant advantages over oral or parenteral administration. Azmacort® (triamcinolone acetamide), Ventolin® HFA (albuterol sulfate), and Serevent® (salmeterol) are examples of commercially available aerosols for the treatment of asthma.
Inserts, Implants, and Devices
Inserts, implants, and devices are used to control drug delivery for localized or systemic drug effects. In these systems, drugs are embedded into biodegradable or nonbiodegradable materials to allow slow release of the drug. The inserts, implants, and devices are inserted into a variety of cavities (e.g. vagina, buccal cavity, cul de sac of the eye, or subcutaneous tissue). A number of degradable and nondegradable inserts are currently available for ophthalmic delivery. These ophthalmic inserts can be insoluble, soluble, or bioerodible. Insoluble inserts are further classified as diffusional, osmotic, and contact lens.
Diffusional and osmotic systems contain a reservoir that is in contact with the inner surface of a controller to which it supplies the drug. The reservoir contains a liquid, a gel, a colloid, a semisolid, a solid matrix, or a carrier containing drug. Carriers consist of hydrophilic or hydrophobic polymers. Degradable inserts consist of polyvinyl alcohol, hydroxypropylcellulose, polyvinylpyrrolidone, and hyaluronic acid. Nondegradable inserts are prepared from insoluble materials such as ethylene vinyl acetate copolymers and styrene-isoprene- styrene block copolymers. The initial use of contact lenses was for vision correction; however, they are becoming more useful as potential drug delivery 18devices by presoaking them in drug solutions. The use of contact lenses can simultaneously correct vision and release drug.
Transdermal patches deliver drugs directly through the skin and into the bloodstream. In general, patches are composed of three key compartments: A protective seal that forms the external surface and protects it from damage, a compartment that holds the medication itself and has an adhesive backing to hold the entire patch on the skin surface, and a release liner that protects the adhesive layer during storage and is removed just prior to application. Watson Pharmaceuticals, Inc. has developed Alora® (estradiol) and Androderm® (testosterone) transdermal patches for the treatment of menopausal symptoms and endogenous testosterone deficiency, respectively.
An intrauterine device is a birth control device placed in the uterus, also known as an IUD or a coil (this colloquialism is based on the coil-shaped design of early IUDs). The IUD is the world's most widely used method of reversible birth control, currently used by nearly 160 million women (just over two-thirds of whom are in China where it is the most widely used birth control method, surpassing sterilization). The device has to be fitted inside or removed from the uterus by a doctor or qualified medical practitioner. It remains in place the entire time pregnancy is not desired. Depending on the type, a single IUD is approved for 5 to 10 years, and trials have demonstrated the copper T 380A to be effective for at least 12 years.
PHARMACEUTICAL INGREDIENTS
To turn a drug substance into a pharmaceutical dosage form or a drug delivery system, pharmaceutical ingredients are required. For example, in the preparation of tablets, diluents or fillers are commonly added to increase the bulk of the formulation. Binders are added to promote adhesion of the powdered drug to other ingredients. Lubricants assist the smooth tabletting process. Disintegrants promote tablet break-up after administration. Coatings improve stability, control disintegration, or enhance appearance. Similarly, in the preparation of pharmaceutical solutions, preservatives are added to prevent microbial growth, stabilizers are added to prevent drug decomposition, and colorants and flavorants are added to ensure product appeal. Thus for each dosage form, the pharmaceutical ingredients establish the primary features of the product and control the physicochemical properties, drug release profiles, and bioavailability of the product. Table 1.3 lists some typical pharmaceutical ingredients used in different dosage forms.
|
PRINCIPLES OF DOSAGE FORM DESIGN
The principal objective of dosage form design is to achieve a predictable therapeutic response to a drug included in a formulation which is capable of large scale manufacture with reproducible product quality. To ensure product quality, numerous features are required-chemical and physical stability, suitable preservation against microbial contamination, uniformity of drug dose, acceptability to prescriber and patient as well as packaging and labeling. Ideally dosage forms should be independent of patient to patient variation. In recent years increasing attention has been directed towards eliminating variation in bioavailability characteristics for chemically equivalent products. It is now recognized that formulation factors can influence therapeutic response of a drug.
There are numerous dosage forms into which a drug substance can be incorporated for the convenient and efficacious treatment of a disease. Dosage forms can be designed for administration by alternative delivery routes to maximize therapeutic response. Table 1.4 lists the range of dosage forms that can be used to deliver drugs by various routes of administration. Many drugs are formulated into several dosage forms of varying strength, each having selected pharmaceutical characteristics suitable for a specific application.
A rational approach to dosage form design for any drug requires complete understanding of its physicochemical and biopharmaceutical properties and therefore dictates the selection and formulation of dosage form are:
|
- Its partition coefficient between lipoidal barriers and aqueous physiological media.
- Its stability and/or degradation rate in physiological fluids.
- Its susceptibility to metabolic inactivation
- Its mechanism of transport through biological membranes.
Biopharmaceutical Aspects of Dosage Form Design
The absorption, distribution, biotransformation and elimination of a drug from the body are dynamic processes that continue from the time a drug is taken until all of the drug has been removed from the body. Once a drug is administered and drug absorption begins, the drug does not remain in a single body location, but rather is distributed throughout the body until its ultimate elimination. For instance, following the oral administration of a drug and entry into the gastrointestinal tract, a portion of the drug is absorbed into the circulatory system from which it is distributed to the various other body fluids, tissues and organs. From these sites the drug may return to the circulatory system and be excreted through kidney as such or the drug may be metabolized by liver or other cellular sites and be excreted as metabolites. Drugs administered by intravenous injection are placed directly into circulatory system avoiding the absorption process which is required from all other routes of administration for systemic effects.
Drugs are absorbed in two general ways, by passive diffusion and through specialized transport mechanism. The passive diffusion is used to describe passage of drug molecules through a membrane which behaves inertly in that it does not actively participate in the process. In passive diffusion the absorption process is driven by the concentration gradient existing across the membrane, with the passage of drug molecules occurring from side of high drug concentration. Lipid solubility and degree of ionization of drug at the absorbing site influence the rate of diffusion. Several specialized transport mechanisms are postulated, including active and facilitated transport. Once absorbed, the drug can exert therapeutic effect either locally or at a site of action remote from that of administration. In latter case the drug has to be transported in body fluids.
The absorption pattern of drugs varies considerably between individual substances as well as between the different administration 22routes. Dosage forms are designed to provide the drug in a suitable form for absorption from each selected route of administration. When the drug is administered from dosage forms designed to deliver via the buccal, respiratory, rectal intramuscular or subcutaneous routes, it passes directly into blood-stream from absorbing tissues, but intravenous is the most direct of all. The physical form of the oral dosage form will also influence absorption rate and onset of action, with solutions acting faster than suspensions, which in turn generally act faster than capsules and tablets.
Drug Factors in Dosage Form Design
Each type of dosage form requires careful study of the physical and chemical properties of drug substances to achieve a stable, effective product. These properties, such as dissolution, crystal size and polymorphic form, solid-state stability and drug-additive interactions, can have profound effects on the physiological availability and physical and chemical stability of the drug. By combining such data with those from pharmacological and biochemical studies, the most suitable drug form and additives can be selected for the formulation of chosen dosage forms.
The process by which a drug particle dissolves is termed dissolution. As a drug particle undergoes dissolution, the drug molecules on the surface are first to enter into solution creating a saturated layer of drug-solution which envelops the surface of solid drug particle. This layer of solution is referred to as diffusion layer. From this diffusion layer, the drug molecules pass throughout the dissolving fluid and make contact with biological membranes and absorption ensues. As the molecule of drug continue to leave diffusion layer, the layer is replenished with dissolved drug from the surface of drug particle and the process of absorption continues. If rate of dissolution for a drug particle is slow, as may be due to physiochemical characteristics of the drug substance or the dosage form, the dissolution process itself would be rate-limiting step in the absorption process.
Particle size reduction results in increase in specific area of powders. Drug dissolution rates, absorption rate, content uniformity in dosage form and stability all are dependent on particle size, size distribution and interaction of solid surfaces. Micronization of poorly aqueous soluble drugs like griseofulvin, tolbutamide, indomethacin, spironolactone and nifidipine has resulted in superior dissolution rates and higher bioavailability. Dosage forms affected by particle size include tablets, 23capsules, powders, suspensions, inhalation aerosols and topical formulations.
All drugs should possess at least limited aqueous solubility for therapeutic efficiency. Solubility and degree of saturation in the vehicle is important in absorption of drugs. Most drugs are either weak acids or weak bases. The solubility of acidic or basic compounds is pH dependent. One of the easiest approaches to enhance solubility and dissolution rate of such drugs is to convert them into their salt forms. With weakly acidic drugs a strong base salt is prepared and in case of weakly basic drugs a strong acid salt is prepared. Alternatively micronizing, complexation or solid dispersion techniques might be employed. Poor aqueous solubility is not always a limitation; it may be desirable feature for a sustained effect after oral or parenteral administration.
The absorbing membrane acts as a lipophilic barrier to the passage of drugs which is related to the lipophilic nature of drug molecule. The partition coefficient, for example between oil and water, is a measure of lipophilic character. The lipid/aqueous partition coefficient of a drug molecule affects its absorption by passive diffusion. In general octanol/pH 7.4 buffer partition coefficients in the range of 1 to 2 are sufficient for absorption across lipoidal membranes.
The amorphous or crystalline character of a drug substance may be of considerable importance to its ease of formulation and handling, its chemical stability and biological activity. Since the amorphous form of a drug is more soluble than crystalline form, different extents of drug absorption may result with consequent differences in degree of pharmacologic activity obtained from each. Some drugs that exist in crystalline form are capable of forming different types of crystals and this property whereby a single chemical substance may exist in more than one crystalline form is known polymorphism. The various polymorphic forms of the same drug differ in many physical and chemical properties including the rate and extent of absorption into the body system. The use of metastable forms generally results in higher solubility and dissolution rates than the respective stable crystal forms of the same drug. On the other hand, the stable polymorph is generally more resistant to chemical degradation. In general the advantages of the metastable crystalline forms in terms of increased physiologic availability of the drug must be balanced against the increased product stability when stable polymorphs are employed.
24Compounds that are intended for oral administration and can undergo rapid degradation at low pH may require protection from acidic environment of the stomach. Protection can often be afforded by administering the drug in the form of an acid-insoluble chemical species or in a dosage form with an acid-resistant coating. Metabolic inactivation of a compound following oral administration can occur in GI lumen, the GI mucosa or the liver. The site of metabolism and the susceptibility of the metabolic processes to saturation are factors that may influence oral bioavailability.
Therapeutic Considerations in Dosage Form Design
The nature of clinical indication, disease or illness against which the drug is intended is an important factor when selecting the range of dosage forms to be prepared. In the vast majority of cases a single drug substance is prepared into a number of dosage forms to satisfy both the particular preferences of the patient or physician and the specific needs of a certain clinical situation. Patients requiring urgent relief from angina pectoris, a coronary circulatory problem, place tablets of nitroglycerin sublingually for rapid drug absorption from buccal cavity. Local effects are generally restricted to dosage forms applied directly such as those applied to the skin, ear, eye and throat. Some drugs may be well absorbed by one route and not another, and must therefore be considered individually.
The age of patient also plays a role in defining the types of dosage forms made available. Infants generally prefer liquid dosage forms, usually solutions and mixtures given orally. Also with liquid preparation the amount of drug administered can be readily adjusted by dilution to give the required dose for particular patient taking weight, age and patient's condition into account. Children can have difficulty in swallowing solid dosage forms and for this reason many oral preparations are prepared as pleasantly flavored syrups or mixtures. Adults generally prefer solid dosage forms, primarily because of their convenience. Interest has grown in the design of formulations that deliver drugs to specific targets in the body, for example the use of liposomes and nanoparticles, as well as providing drugs over longer periods of time at controlled rates.
APPROACHES IN DOSAGE FORM DESIGN
Once the physicochemical and biopharmaceutical properties of the drug are determined and desired plasma concentration profile 25is defined, the pharmaceutical scientist can select and develop an efficacious dosage form by utilizing a formulation approach, a prodrug approach, a device approach or an alternative administration route approach.
Formulation Approach
With the use of formulation techniques the bioavailability could be improved and/or toxicity and side effects of drugs are reduced. Several factors including particle size, crystalline habit and salt form can affect the solubility and dissolution rate. The effect of particle size on dissolution rate of relatively insoluble compounds becomes significant when the drug is administered in an uncompressed form such as suspension or capsule and the material is well dispersed within the GI tract.
Polymorphism is the ability of a chemical species to crystallize in more than one distinct crystal habit. The differences in dissolution rate and solubility that polymorphs can produce may have a dramatic impact on bioavailability when dissolution is the rate-limiting step in the absorption process. The selection of salt form directly influences the physicochemical and biopharmaceutical properties of a compound. For highly insoluble amine bases such as ergotamine and certain antimalarials, the drug may precipitate in the small intestine as soon as the pH rises following stomach emptying. Thus the selection of most appropriate salt form should be made early in the development process to optimize bioavailability.
The stability of a drug in the gut is influenced by both chemical and enzymatic factors. Protection from chemical degradation may be accomplished via coating techniques and enzymatic protection may be achieved with enzymatic inhibitors. An enteric coating protects the drug during transit through the acidic medium of the stomach. Upon entering the higher pH environment of the duodenum, the coating is dissolved and the drug becomes available for absorption. Such a coating also provides protection for the gut mucosa when the drug is capable of producing GI irritation. The limitations of enteric coating dosage forms include the possibility of duodenal irritation from caustic drugs, and an increase in intersubject variability due to presentation to the small intestine of a dosage form that needs to undergo disintegration and dissolution versus a disintegrated and partially dissolved drug substance.
Enzyme inhibitors compete with the active drug for the enzyme and thereby reduce the degradation of the drug and deliver, it more 26efficiently to the systemic circulation. Alternatively the enzyme in gut can be utilized to control the release of the active drug in the gut.
The mechanism of absorption must always be evaluated, when a sustained release dosage form is considered. A drug, that is passively absorbed throughout the GI tract is an ideal candidate for sustained release. Drugs which have windows of absorption due to site-specific or active transport process may have incomplete bioavailability when formulated in oral sustained-release dosage forms.
Prodrug Approach
A prodrug is a chemically modified inert drug precursor, which upon biotransformation liberates pharmacologically active parent compound. An ideal prodrug should possess the following properties:
- It should not have intrinsic pharmacologic activity
- It should rapidly transform, chemically or enzymatically into active form when desired
- The metabolic fragments, apart from the active drug should be non-toxic
- It should be eliminated more slowly than its rate of cleavage to the parent
- It should be inexpensive.
Prodrug can be used to increase or decrease the aqueous solubility, mask bitterness, increase lipohilicity, improve absorption, decrease local side-effects and alter tissue distribution of the parent molecule. The various applications of this approach are given in Table 1.5.
Hydrophilic drugs are desired where dissolution is the rate-limiting step in absorption of poorly soluble aqueous agents or when parenteral or ophthalmic formulation of such agents is desired.
|
27Drugs with hydroxyl function can be converted into their hydrophilic forms by use of half-esters such as hemisuccinates, hemiglutarates or hemiphthalates; the other half of these acidic carriers can form sodium, potassium or amine salts and render the moiety water soluble.
The perception of taste requires that some minimum aqueous concentration be exceeded so that the taste can be detected. As a result bitterness can be masked by reducing solubility in saliva. Some drugs, which are in liquid form, are unsuitable for formulation as a tablet especially if their dose is high. The method of converting such liquid drugs into solid prodrugs involves formation of symmetrical molecules having a higher tendency to crystallize.
Membrane permeability is governed in part by the lipohilicity of a compound. Highly polar compounds have low lipohilicities and therefore low membrane permeability. The lipophilic form of a drug has enhanced membrane/water partition coefficient as compared to the hydrophilic form, thus favoring passive diffusion. The bioavailability of topically administered drugs also depends upon lipid solubility. Skin permeability of polar drugs can be improved by esterification to form lipid soluble compounds.
Altering the tissue distribution of a compound to transport the drug molecule to the site of action is an important delivery method. This method can produce striking results when the site of action is the brain and the blood-brain barrier must be crossed. A more recent novel approach for delivery of drugs to brain is the use of dihydropyridine-pyridinium salt redox system. A prodrug can also be designed to selectively target such tumorous cells where it can be activated to parent antineoplastic agent.
Device Approach
The controlled release systems are so designed that they release the drug over a prolonged period of time usually longer than the typical dosing interval for a conventional formulation. The rationale of a controlled release drug delivery system is to optimize the biopharmaceutic, pharmacokinetic and pharmacodynamic properties of a drug in such a way that its utility is maximized through reduction in side-effects and treat or control of condition in the shortest possible time by using small quantity of drug administered by the most suitable route. The application of this approach includes transdermal patches, ocusert, intrauterine devices, etc.
28In diffusion controlled devices, the rate-controlling step is not the dissolution but the diffusion of dissolved drug through polymeric barrier. The two types of diffusion controlled systems are: Matrix systems and reservoir devices. In matrix systems the drug is dispersed randomly in an insoluble polymer, whereas reservoir devices surround the drug with an intact rate-controlling membrane.
Pilocarpine ocusert is a contact lens-shaped device that is inserted into the lower cul-de-sac of the eye. It provides continuous release of pilocarpine at rates of 10 or 20 mcg per hour over a period of 7 days for the treatment of open-angle glaucoma.
The progestasert intrauterine device is a white, T-shaped polyethylene device with progesterone reservoir dispersed in a silicone polymer placed in the vertical arm, which is enclosed in a sleeve of rate-controlling membrane of ethylene-vinyl acetate copolymer. The device releases progesterone at a rate of 65 mcg/day for a period of one year.
Alternative Administration Routes
An individual drug substance may be formulated into multiple dosage forms which result in different drug absorption rates and times of onset, peak and duration of action. The difference in drug absorption between dosage forms is a function of the formulation and the route of administration.
Administration of drugs by alternative route avoids absorption and metabolic barriers that may be present in GI tract. The routes can also provide systemic availability when oral administration is contraindicated due to physiological condition, or the route may provide for a concentration-time profile that approaches intravenous dosing profiles. The transdermal, ophthalmic, nasal, buccal and rectal route each provides one or more of these advantages.
Ophthalmic route has traditionally been used for topical application for local effects. Current research is directed towards the feasibility of nasal administration of peptides for systemic drug delivery. After topical administration in the eye, peptides can be absorbed from the mucosa during tear turnover as well as via the blood vessels of conjunctiva. Nasal administration produces rapid blood levels and rapid responses that approach those obtained from intravenous dosing with many drugs such as propranolol hydrochloride, barbiturates, benzoic acid, etc. Although many drugs are absorbed rapidly and quantitatively following nasal administration, peptides have generally shown low bioavailabilities.
29The buccal and sublingual routes of administration permit rapid delivery to the systemic circulation. Absorption from buccal and sublingual vasculature and lymphatics bypasses hepatic circulation and thereby reduces first-pass metabolism. Organic nitrates and testosterone have been administered by these routes to produce rapid plasma concentrations and to minimize hepatic metabolism.
Transdermal administration avoid first-pass metabolism and provide a large surface area for continuous controlled administration of drugs with short biological half-lives and narrow therapeutic indices. The route has been used for a numbers of drugs for systemic delivery including nitroglycerin, clonidine, estradiol, scopolamine, etc.
The lungs provide an excellent surface for absorption when the drug is delivered in gaseous, aerosol mist or ultrafine solid particle form. This delivery route is being explored as a means of administering the therapeutic agents emerging from biotechnology such as proteins and peptides.
Rectal administration for systemic effect has traditionally been limited to clinical situations where oral intake is restricted due to physiological condition. Rectal route of administration is indicated for drugs that are inactivated by GI fluids or inactivated by the liver following oral absorption. The route has been utilized for aspirin, acetaminophen, aminophylline, promethazine, chlorpromazine and indomethacin. The rectal administration of drugs is limited due to increased interpatient variability and patent acceptability.
DRUG DEVELOPMENT AND UNITED STATES FDA
In the US, all food, drugs, cosmetics and medical devices for both humans and animals are regulated under the authority of Food and Drug Administration (FDA). Discovering and developing safe and effective new medicines is a long, difficult, and expensive process. The research-based pharmaceutical industry expects to invest $24 billion in research and development in 1999. Once a new compound has been identified in the laboratory, it undergoes the drug development process before it can be marketed for public use. According to US FDA, the entire drug development process can be summarized in a series of steps:
- Preclinical testing
- Investigational new drug application (INDA)
- Clinical trials
- New drug application (NDA)
- NDA approval.
30Preclinical testing: The compound is tested for safety and efficacy in laboratory and animal studies. Preformulation studies covering the physical and chemical characterization of the new drug substance are also conducted. Preclinical testing involves:
- Pharmacology
- Toxicology
- Preformulation
- Formulation
- Analytical
- Pharmacokinetics.
During non-clinical development, experts such as pharmacologists and toxicologists carry out in vitro and in vivo tests to ascertain the safety and prove the pharmacological efficacy of the compound. The studies performed provide information on the mechanisms of action resulting in a pharmacological effect, and how the new drug interacts with the systems of living bodies. In particular the tests are designed to detect unwanted side effects of administration, either pharmacological or toxicological. This data is important for determining if the new drug is safe for clinical administration, and also provides information on dosage and route of administration.
Formulation developments starts immediately after or concurrently with preformulation studies. One of the first steps after selecting an active pharmaceutical ingredient (API) for development is to find the best way of delivering the molecule. Knowledge around delivering technologies resides within the pharmaceutical formulation department. The formulation work is usually divided into two phases—early and late stage formulation.
Early stage formulation focuses on producing simple, but suitable products for use in toxicological and early clinical testing. In parallel the API undergo a series of physical and chemical testing to learn about the stability of the API under different conditions.
Depending on the dosage form selected, various studies are conducted to accomplish the goal. Formulations developed are tested for both physical and chemical stability at accelerated temperatures. To determine the chemical stability of the formulations, an analytical method needs to be in place (usually an HPLC method) to identify and quantify the active compound. Care should be taken to see that the method is selective and specific for the active molecule (Flow chart 1.1).
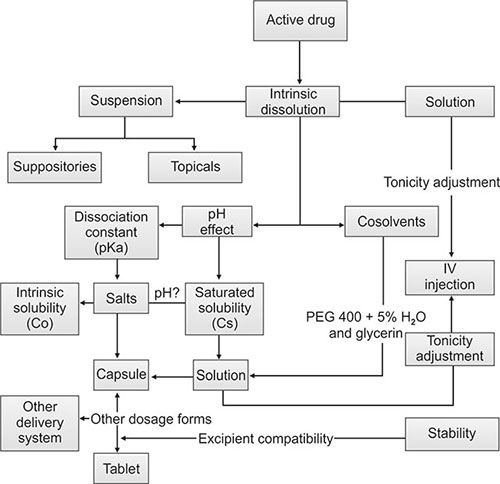

Late stage formulation work focuses on fine tuning the formulation that eventually will be the final medical product to be marketed. In between the formulation will have undergone development from bench scale to pilot scale and finally to production scale, known as process development. In parallel the final formulation will undergo extensive long term stability testing.
It cannot be stressed too much that an early assessment of the desired dosage-form properties can contribute greatly to the speed of the drug development process. The lack of either an appropriate dosage form or a validated manufacturing process during the clinical trial can result in disaster.
Pharmaceutical analysis is a support function for pharmaceutical formulation, which means that the two departments have to work very closely with each other. The main focus in pharmaceutical analysis is developing the necessary analytical methods to confirm 32the chemical structure of the API, its content, purity and quality and the pharmaceutical formulations used during the development of the medical product from early stage to hand-over to quality control in production.
Investigational new drug application: After completing preclinical testing, a company files an IND with food and drug administration (FDA) to begin to test the drug in people. The IND enables a sponsor to ship an unapproved drug in interstate commerce. Clinical trials may proceed 30 days after filing unless the FDA places a hold on the proposal in USA (Flow chart 1.2).
Clinical trials: Clinical development is the phase of the product development stage when the potential new drug is subjected to detailed clinical testing which involves human clinical trials (Table 1.6).

|
Phase I
- Small group of healthy volunteers
- Evaluate drug safety
- Determine a safe dosage range
- Identify side effects
- Collect data for pharmacokinetic analysis.
Phase II
- Larger group of people with disease
- Evaluate drug effectiveness
- Further evaluate drug safety
- Further evaluate dosage.
Phase III
- Large groups of people with disease
- Confirm drug's effectiveness
- Monitor side effects
- Compare drug to commonly used treatments
- Monitor interactions with other drugs
- Multicenter trial—many hospitals.
Phase IV
- After the drug has been marketed
- Information about drug effect in various populations
- Side effects associated with long-term use
- New indications: Potential to extend patent protection.
New drug application: Following the completion of all three phases of clinical trials, a company analyzes all of the data and files an NDA with 34FDA if the data successfully demonstrate both safety and effectiveness. The NDA contains all of the scientific information that the company has gathered. NDAs typically run 100,000 pages or more. By law, FDA is allowed six months to review an NDA. The average NDA review time for new molecular entities approved in 1997 was 16.2 months (Flow chart 1.3).
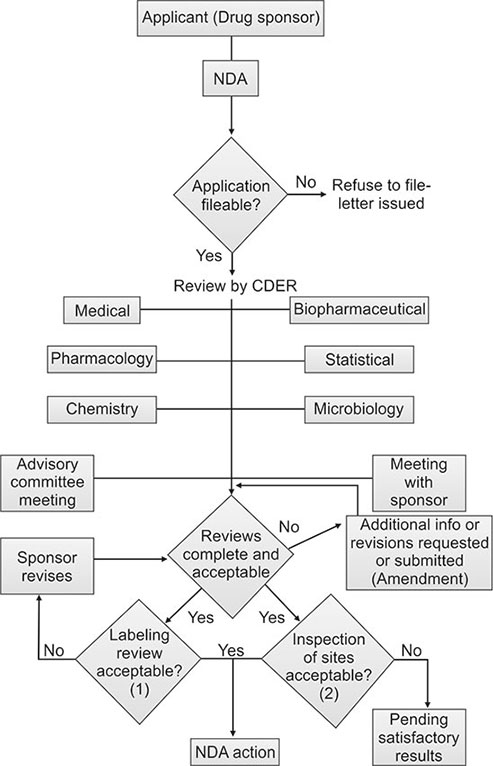
35NDA approval: Once US FDA approves an NDA, the new medicine becomes available for physicians to prescribe. A company must continue to submit periodic reports to FDA, including any cases of adverse reactions and appropriate quality-control records. For some medicines, FDA requires additional trials (Phase IV) to evaluate long-term effects.
BIBLIOGRAPHY
- Ansel HC, Popovich NG, Allen LV. Pharmaceutical Dosage Forms and Drug Delivery Systems, 6th edition (1st Indian edition). BI Waverly Pvt. Ltd. 1995;pp.21–150.
- Billany M. Suspensions and emulsions. In: Pharmaceutics: Aulton ME (ed), The Science of Dosage Form Design, 2nd edition. Churchill Livingstone. 2002;pp.334–59.
- Block LH, Yu ABC. Drug development in the Pharmaceutical Industry. In: Comprehensive Pharmacy Review, Shargel L, Mutnick AH, Souney PF, Swanson LN (eds). Lippincott Williams and Wilkins. 2001;pp.1–8.
- Boylan JC. Liquids. Lachman L, Lieberman HA, Kanig JL (eds), In: The Theory and Practice of Industrial Pharmacy, 3rd edition. Varghese Publishing House. 1991;pp.457–75.
- Boylan JC, Nail SL. Parenteral products. Banker GS, Rhodes CT (eds), In: Modern Pharmaceutics, 4th edition. Marcel Dekker, Inc. 2002;pp.381–414.
- Cartensen JT. Pharmaceutics of Solids and Solid Dosage Forms. John Wiley and Sons. 1976;pp.1–52.
- Carter SJ. Cooper and Gunn's Dispensing for Pharmaceutical Students, 12th edition. CBS Publishers and Distributors. 1987;pp.8–12,634–62.
- Florence AT, Attwood D. Physicochemical Principles of Pharmacy, 3rd edition Palgrave, MacMillan. 1998;pp.11–76.
- Gupta P, Garg S. Recent advances in semisolid dosage forms for dermatological application. Pharm. Tech. 2002;144–62.
- Hadgraft JW. Rectal administration. Rawlins EA (ed), In: Bentley's Textbook of Pharmaceutics, 8th edition. Bailliere Tindall. 1992;pp.347–9.
- Hillery AM. Advanced drug delivery and targeting: An introduction. Hillery AM, Lloyd AW, Swarbrick J (eds). In: Drug Delivery and Targeting: For Pharmacists and Pharmaceutical Scientists, Taylor and Francis. 2001;pp.63–82.
- Indian Pharmacopoeia, Vol. I and II. The Controller of Publication. 1996;pp.134–46,734–6.
- Jackson WA. From electuaries to enteric coating: a brief history of dosage forms. Anderson S (ed). In: Making Medicines: A Brief History of Pharmacy and Pharmaceuticals, Pharmaceutical Press, London. 2005;pp.1–50.
- Jain NK, Sharma SN. A Textbook of Professional Pharmacy, 3rd edition. Vallabh Prakashan. 1994;pp.1–13, 237–59.
- Kohli DPS, Shah DH. Drug Formulations Manual. 2nd edition. Eastern Publishers. 1998;pp.4–10.
- Lieberman HA, Rieger MM, Banker GS. Pharmaceutical Dosage Forms: Disperse Systems, Vol.1. 1988;pp.199–203,217–19,232–6.
- Lionberger RA. FDA critical path initiatives: opportunities for generic drug. The AAPS Journal. 2008;10(1):103–9.
- Machin D. General issues. Machin D, Day S, Green S, Everitt B, George S (eds). In: Textbook of Clinical Trials, John Wiley and Sons, Inc. 2004; pp.11–44.
- Mahato RI. Dosage forms and drug delivery systems. Gourley DR, Eoff JC (eds). In: APhA'S Complete Review for Pharmacy. Castle Connolly Graduate, Medical Publishing, Ltd. 2004;pp.37–63.
- Mahato RI. Pharmaceutical Dosage Forms and Drug Delivery. CRC Press. 2007;pp.1–290.
- Manso PJ, Sokol AL. Life cycle management of ageing pharmaceutical assets. Pharmaceutical Law Insight. 2007;16–9.
- Maurin MB, Hussain AA, Dittert LW. Dosage form design: a physicochemical approach. Swarbrick J, Boylan JC (eds). In: Encyclopedia of Pharmaceutical Technology, Vol.4, Marcel Dekker, Inc. 1991;pp.193–207.
- Mithal BM. A Textbook of Pharmaceutical Formulation, 4th edition. Vallabh Prakashan. 1991;pp.133–60.
- Singh H. Pharmacopoeia and Formularies. Vallabh Prakashan. 1994;pp.1–59.
- Sinko PJ. Martin's Physical Pharmacy and Pharmaceutical Sciences, 5th edition. Lippincott Williams and Wilkins. Indian edition: BI Publications Pvt. Ltd. 2006;pp.1–20.
- Tripathi KD. Essentials of Medical Pharmacology, 5th edition. Jaypee Brothers Medical Publishers (P) Ltd. 2003;pp.3–10.
- Winfield AJ. (original author Moody MM). Routes of administration and dosage forms. Winfield AJ, Richards RME (eds). In: Pharmaceutical Practice, Churchill Livingstone. 2004;pp.174–80.