

INTRODUCTION
Definition: Pathology is the scientific study (logos) of disease (pathos). It mainly focuses on the study of the structural, biochemical and functional changes in cells, tissues and organs in disease.
Learning Pathology
Study of pathology can be divided into general pathology and systemic pathology.
- General pathology: It deals with the study of mechanism, basic reactions of cells and tissues to abnormal stimuli and to inherited defects.
- Systemic pathology: This deals with the changes in specific diseases/responses of specialized organs and tissues.
Scientific Study of Disease
Disease process is studied under following aspects.
Etiology
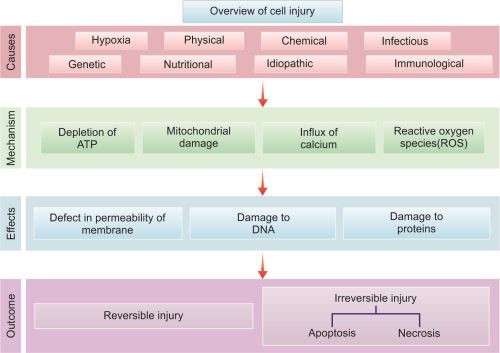
The etiology of a disease is its cause. The causative factors of a disease can be divided into two major categories: Genetic and acquired (e.g. infectious, chemical, hypoxia, nutritional, physical). Most common diseases are multifactorial due to combination of causes.
Pathogenesis
It refers to the sequence by which the causative factor/s produces cellular, biochemical and molecular abnormalities following the exposure of cells or tissues to an injurious agent. Pathogenesis deals with sequence of events that occur in the cells or tissues from the beginning of any disease process. With the present advances in technology, it is possible to identify the changes occurring at molecular level and this knowledge is helpful for designing new therapeutic approaches.
- Latent period: Few causative agents produce signs and symptoms of the disease immediately after exposure. Usually, etiological agents takes some time to manifest the disease (e.g. carcinogenesis) and this time period is called as the latent period. It varies depending on the disease.
- Incubation period: In disorders caused by infectious (due to bacteria, viruses, etc.) agents, the period between exposure and the development of disease is called the incubation period. It usually ranges from days to weeks. Most of the infectious agents have characteristic incubation period.
Molecular Pathology
Most of the diseases can be diagnosed by the morphological changes in tissues. But, with the present advances in diagnostic pathology, the diseases can be analyzed by the molecular and immunological approaches. Molecular pathology has revealed the biochemical basis of many diseases, mainly congenital disorders and cancer. These techniques can detect changes in a single nucleotide of DNA. In situ hybridization can detect the presence of specific genes or their messenger RNA in tissue sections or cell preparations. Minute quantities of nucleic acids can be amplified by the use of the polymerase chain reaction. DNA microarrays can be used to determine patterns of gene expression (mRNA).
Functional Derangements and Clinical Manifestations
- Functional derangements: The effects of genetic, biochemical and structural changes in cells and tissues are functional abnormalities. For example, excessive secretion of a cell product (e.g. nasal mucus in the common cold); insufficient secretion of a cell product (e.g. insulin lack in diabetes mellitus).
- Clinical manifestations: The functional derangements produce, clinical manifestations of disease, namely symptoms and signs. Diseases characterized by multiple abnormalities (symptom complex) are called syndromes.
- Prognosis: The prognosis forecasts (predicts) the known or likely course (outcome) of the disease and, therefore, the fate of the patient.
- Complications: It is a negative pathologic process or event occurring during the disease which is not an essential part of the disease. It usually aggravates the illness. For example, perforation and hemorrhage are complications which may develop in typhoid ulcer of intestine.
- Sequelae: It is a pathologic condition following as a consequence of a disease. For example, intestinal obstruction following healed tuberculosis of intestine, mitral stenosis following healed rheumatic heart disease.
- Remission and relapse:
- Remission: It is the process of conversion from active disease to quiescence. Some of the chronic diseases are interspersed by periods of quiescence when the patient is relatively in good health.
- Relapse: It is the process in which the signs and symptoms of disease reappear.
Some diseases may pass through several cycles of remission and relapse. For example, inflammatory bowel disease (Crohn's disease and ulcerative colitis).
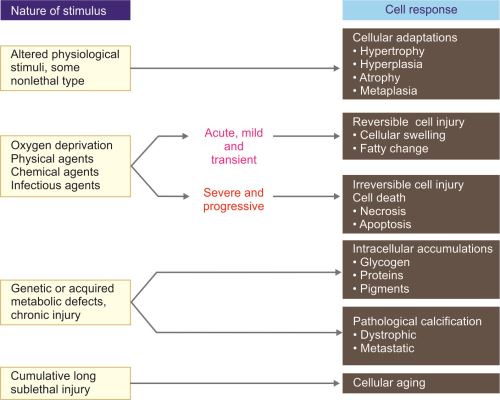
TYPES OF CELLULAR RESPONSES TO INJURY
Depending on the nature of stimulus/injury, the cellular responses can be mainly divided into four types (Fig. 1.1).
- Cellular adaptations
- Cell injury
- Reversible cell injury
- Irreversible cell injury.
- Intracellular accumulations
- Pathologic calcification.
Different stages of cellular responses to stress and injurious stimuli are shown in Figure 1.2.


CELLULAR ADAPTATIONS
Q. Write short note on cellular adaptations.
When the cell is exposed to pathological stimuli, the cells can achieve a new, steady altered state that allows them to survive and continue to function in an abnormal environment. These are reversible changes in the size, number, phenotype, metabolic activity or functions of cells constitute cellular adaptations.
Types of adaptations: Hypertrophy, hyperplasia, atrophy and metaplasia.
Hypertrophy
Definition: Increase in the size of the tissue or organ due to increase in the size of cells.
Causes
Increased functional demand/workload.
Physiological
- Hypertrophy of skeletal muscle: For example, the bulging muscles of body builders and athletes.
- Hypertrophy of smooth muscle: For example, growth of the uterus during pregnancy from estrogenic stimulation.
Pathological
- Hypertrophy of cardiac muscle: For example, left ventricular hypertrophy (Fig. 1.3) due to hypertension or damaged valves (aortic stenosis, mitral incompetence).
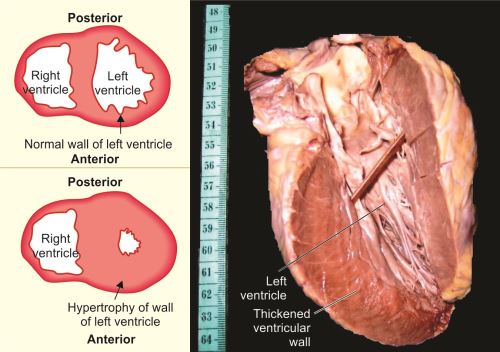
Figs 1.3A to C: (A) Transverse section of normal heart; (B) Transverse section of heart with thickening of wall of the left ventricle due to hypertrophy; (C) Longitudinal section of heart with left ventricular hypertrophy
Mechanisms of Cellular Hypertrophy
Hypertrophy is due to increased synthesis of cellular proteins. Steps involved in biochemical mechanisms of myocardial (cardiac muscle) hypertrophy are shown in Figure 1.4.
Activation of the Signal Transduction Pathways
Various mechanisms involved are:
Physiologic hypertrophy:
Increased workload on the myocardium produces mechanical stretch and is the major trigger for physiological hypertrophy.
Pathologic hypertrophy:
Growth factors and hypertrophy agonists are involved in pathologic hypertrophy.
- Growth factors: These include (TGF-β), insulin-like growth factor-1 (IGF-1) and fibroblast growth factor (FGF).
- Hypertrophy agonists: These include α-adrenergic agonists, endothelin-1, angiotensin II, nitric oxide (NO), and bradykinin.
Mechanical sensors also stimulate production of growth factors and agonists. They cause increased synthesis of muscle proteins.
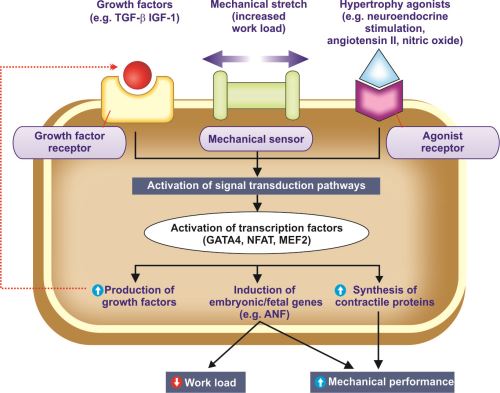
Fig. 1.4: Mechanisms of myocardial hypertrophy.
Abbreviations: ANF, atrial natriuretic factor; IGF-1, insulin-like growth factor, TGF-β, transforming growth factor-β
Activation of Transcription Factors
Mechanical stretch, growth factors and hypertrophy agonists activate the signal transduction pathways and transcription factors [e.g. GATA4, nuclear factor of activated T-cells (NFAT) and myocyte enhancer factor 2 (MEF2)]. The activated transcription factors results in:
- Increased synthesis of contractile proteins: This is necessary to meet the increased functional demand.
- Induction of embryonic/fetal genes: Some genes are normally expressed only during early development of embryo and fetus. They are re-expressed in hypertrophied cells. For example, the gene for atrial natriuretic factor (ANF) is expressed in the embryonic heart, but not expressed after birth. In cardiac hypertrophy, ANF gene is re-expressed. ANF is a hormone that causes salt secretion by the kidney, decreases blood volume and pressure. Its re-expression decreases hemodynamic workload and increases the mechanical performance.
- Increased production of growth factors.
Hyperplasia
Q. Write short note on hyperplasia.
Definition: Increase in the number of cells in an organ or tissue, resulting in increased size/mass of the organ or tissue.
Causes
- Physiological hyperplasia: Hormonal stimulation or as compensatory process.
- Hyperplasia due to hormones: For example, hyperplasia of glandular epithelium of the female breast at puberty, pregnancy and lactation, hyperplasia of the uterus during pregnancy from estrogenic stimulation
- Compensatory hyperplasia: For example, in liver following partial hepatectomy.
- Pathological hyperplasia: Due to excess endocrine stimulation or chronic injury/irritation.
- Excessive hormonal stimulation: For example, endometrial hyperplasia (due to estrogen, refer Figs 23.12 and 23.13) and benign prostatic hyperplasia [due to androgens (Figs 1.5 and 22.3 to 22.5)].
- Chronic injury/irritation: Long-standing inflammation or chronic injury may lead to hyperplasia especially in skin or oral mucosa.
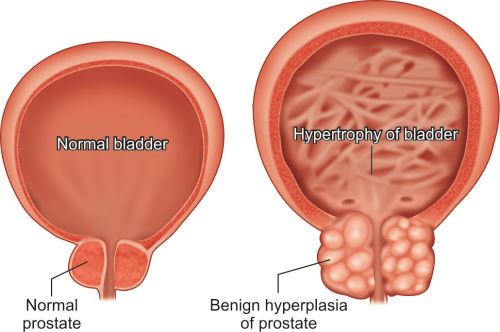
Pathological hyperplasia can progress to cancer, e.g. endometrial hyperplasia can develop into endometrial cancer.
Mechanism
- Hyperplasia is characterized by cell proliferation mostly of mature cell mediated through stimulation by growth factor or hormones.
- In some cases, the new cells may be derived from tissue stem cells.
Atrophy
Q. Write short note on atrophy.
Definition: Atrophy is the reduced size of an organ or tissue resulting from a decrease in cell size and number.
Causes
Physiological atrophy: Common during normal fetal development and in adult life.
- During fetal development: For example, atrophy of embryonic structures such as thyroglossal duct.
- During adult life: For example, involution of thymus, atrophy of brain, gonads and heart due to aging (senile atrophy).
Pathological atrophy: Local or generalized.
- Local
- Disuse atrophy (decreased workload): For example, atrophy of limb muscles immobilized in a plaster cast (as treatment for fracture) or after prolonged bed rest.
- Denervation (loss of innervation) atrophy: For example, atrophy of muscle due to damage to the nerves (e.g. poliomyelitis).
- Ischemic (diminished blood supply) atrophy: For example, brain atrophy produced by ischemia due to atherosclerosis of the carotid artery.
- Pressure atrophy: For example, atrophy of renal parenchyma in hydronephrosis due to increased pressure.
- Generalized
- Starvation (inadequate nutrition) atrophy: For example, protein-calorie malnutrition.
Mechanisms
Atrophic cells have diminished function. There is decreased protein synthesis and increased protein degradation in cells.
Differences between atrophy, hypertrophy and hyperplasia are listed in Table 1.1.
Q. List the differences between atrophy, hypertrophy and hyperplasia.
Metaplasia
Q. Write short note on metaplasia with examples.
Definition: Metaplasia is a reversible change in which one adult cell type is replaced by another adult cell type.
Causes
- Metaplasia is usually fully reversible adaptive response to chronic persistent injury. If the noxious stimulus is removed (e.g. cessation of smoking), the metaplastic epithelium may return to normal.
- Metaplasia is mainly seen in association with tissue damage, repair and regeneration.
- The replacing cell type is usually more suited to a change in environment.
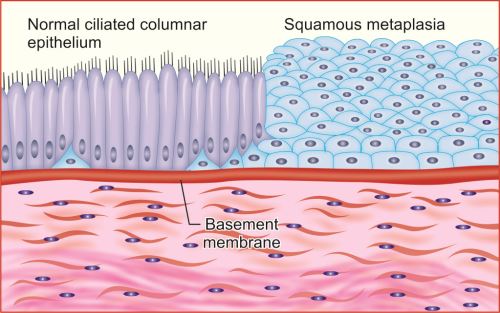
Types of Metaplasia
Epithelial Metaplasia
Squamous metaplasia: Original epithelium is replaced by squamous epithelium.
- Respiratory tract: For example, chronic irritation due to tobacco smoke, the normal ciliated columnar epithelial cells of the trachea and bronchi undergo squamous metaplasia (Fig. 1.6).
-
Columnar metaplasia: Original epithelium is replaced by columnar epithelium.
- Squamous to columnar: In Barrett esophagus, the squamous epithelium of the esophagus replaced by columnar cells (refer Fig. 18.1).
- Intestinal metaplasia: The gastric glands are replaced by cells resembling those of the small intestine.
Connective Tissue Metaplasia
- Osseous metaplasia: Formation of new bone at sites of tissue injury is known as osseous metaplasia. Bone formation in muscle, known as myositis ossificans, occasionally occurs after intramuscular hemorrhage. Other examples include cartilage of larynx and bronchi in elderly individual, scar of chronic inflammation of long duration, fibrous stroma of tumor (e.g. leiomyoma).
Mechanism
Develops due to the reprogramming of precursor cells (i.e. stem cells or undifferentiated mesenchymal cells) that are present in normal tissues.
CELL INJURY
Q. Write short note on causes of cell injury.
Definition: Cell injury is the effect of stresses due to variety of etiological agents on the cell.
Causes of Cell Injury
- Hypoxia: It refers to inadequate oxygenation of tissue. It is the most common cause of cell injury.Causes of hypoxia:
- Decreased blood flow is called ischemia. It may be due to thrombosis, embolism, atherosclerosis or external compression of vessel.
- Inadequate oxygenation of the blood (hypoxemia)
- Due to pulmonary disease.
- Decreased perfusion of tissues: For example; cardiac failure, hypotension shock.
- Decreased oxygen-carrying capacity of the blood: For example, anemia.
- Severe blood loss.Mechanism of injury: Hypoxia causes cell injury by reducing aerobic oxidative respiration and decreasing the synthesis of adenosine triphosphate (ATP).Outcome: Depending on the severity of the hypoxia, cells may adapt, undergo injury, or die.
- Physical Agents:
- Mechanical trauma: For example, blunt/penetrating/crush injuries, gunshot wounds.
- Thermal injury: Extremes of temperature (burns and deep cold).
- Radiation (ionizing radiation and non-ionizing radiation).
- Electric shock.
- Pressure changes: Sudden changes in atmospheric pressure.
- Chemical Agents:
- Heavy metals and poisons: For example, arsenic, mercuric salts or cyanide.
- Simple chemicals: For example, hypertonic concentrations of glucose or salt.
- Strong acids and alkalies.
- Oxygen at high concentrations is toxic.
- Environmental and air pollutants: For example, insecticides, and herbicides.
- Industrial and occupational hazards (carbon monoxide and asbestos).
- Social/lifestyle choices: Addiction to drugs and alcohol, cigarette smoking.
- Therapeutic drugs.
- Infectious Agents: Viruses, bacteria, fungi, rickettsiae and parasites. The mechanism by which these infectious agents cause injury varies.
- Immunologic Reactions
- Autoimmunity: Immune reactions to endogenous self-antigens are responsible for autoimmune diseases.
- Hypersensitivity reactions and other immune reactions: Heightened immune reactions to many external agents (e.g. microbes and environmental agents).
- Genetic Derangements: Genetic defects may cause cell injury because of:
- Accumulation of damaged DNA or misfolded proteins
- Variations in the genetic makeup.
- Nutritional Imbalances:
- Nutritional deficiencies:
- Protein-calorie deficiencies
- Deficiencies of specific vitamins.
- Nutritional excesses:
- Excess of cholesterol predisposes to athero-sclerosis.
- Obesity is associated with increased incidence of several important diseases, such as diabetes and cancer.
- Hypervitaminosis.
- Idiopathic: Cause is not known.
General Principles of Cell Injury
- Cellular response to injury: It depends on: (1) type of injury, (2) duration of injury and (3) severity of injury.
- Consequences of injury: It depends on: (1) type of cell involved, (2) adaptability of cell, (3) status of cell and (4) genetic makeup of the cell.Q. Describe the role of cytosolic calcium in cell injury.
- Targets and biochemical mechanism of cell injury: These include (1) mitochondrial damage/dysfunction, (2) disturbance of calcium homeostasis, (3) damage to cellular membranes and (4) damage to DNA and misfolding of proteins.
Mechanisms of Cell Injury
Q. Write short note on mechanism (biochemical basis) of cell injury.
Injurious stimuli that cause cell injury lead to complex cellular, biochemical and molecular changes. Certain mechanism is common for most forms of cell injury and cell death.
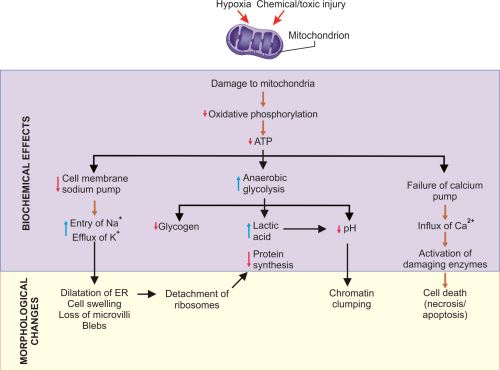
Decreased Production of Adenosine Triphosphate
Adenosine triphosphate (ATP) is required for all processes within the cell. Injury like hypoxia, chemicals (e.g. cyanide) can cause decreased production of ATP.
- Effects of decreased ATP (Fig. 1.7):
- Failure of the cell membrane sodium pump
- Increased anaerobic glycolysis
- Failure of the calcium pump
- Failure of protein synthesis in the ribosomes.

Mitochondrial Damage (Fig. 1.8)
- Mitochondria are sensitive to almost all types of injurious stimuli (e.g. hypoxia, toxins).
Consequences of Mitochondrial Damage
- Depletion of ATP: Its effects are mentioned above.
- Formation of reactive oxygen species (ROS): Its effects are mentioned in page 13 (refer Fig. 1.10).
- Formation of mitochondrial permeability transition pore: It occurs in the mitochondrial membrane. This leads to the loss of mitochondrial membrane potential, pH changes and progressive depletion of ATP and ultimately necrosis of the cell.
- Leakage of mitochondrial proteins into cytoplasm: The mitochondrial membranes contain many proteins such as cytochrome C and proapoptotic proteins (e.g. BAX and BAK). Increased permeability of the mitochondrial membrane may result in leakage of these proteins into the cytosol and induce apoptosis.
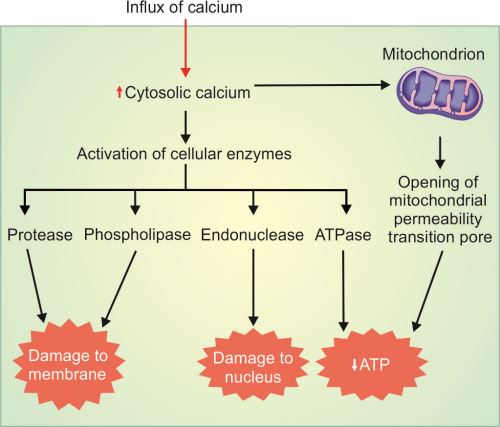
Influx of Calcium and Loss of Calcium Homeostasis (Fig. 1.9)
Normally, concentration of cytosolic calcium is very low and most of it is sequestered in mitochondria and the endoplasmic reticulum (ER). Ischemia and certain toxins cause an increase in cytoslic calcium (Fig. 1.9). Initially, it is due to the release from intracellular stores and later due to influx across the cell membrane. Increased intracellular calcium stimulates activation of several damaging enzymes (e.g. phospholipases, endonucleases and protease) as well as caspases. The net result is apoptosis.
Accumulation of Oxygen-derived Free Radicals (Oxidative Stress)
Q. Write short essay/note on free radical injury and its role in cell injury.
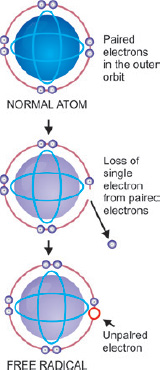
Free radicals are unstable chemical compounds with a single unpaired electron in an outer orbit (Fig. 1.10).
- Normally, free radicals produced in the cells are unstable and are rapidly destroyed.
- When free radicals react with any molecules they convert those molecules into free radicals and thus initiate autocatalytic reactions.
- Excess of free radicals may be either due to increased production or ineffective degradation.

Types of Free Radicals
Q. Write short note on free radical injury.
- Oxygen-derived free radicals: Reactive oxygen species (ROS) are oxygen-derived free radicals. ROS includes superoxide anion (O−•2), hydrogen peroxide (H2O2) and hydroxyl ions (−OH).
- Reactive nitrogen species/nitric oxide derived free radicals: For example, nitric oxide (NO) is generated by endothelial cells (refer Fig. 2.6), macrophages, neurons, and other types of cells. NO can act as a free radical and can also be converted to highly reactive peroxynitrite anion (ONOO−), NO2 and NO3−.
- Free radicals from drug and chemical: Enzymatic metabolism of exogenous chemicals or drugs can generate free radicals which are not ROS but have similar effects (e.g. CCl4 can generate CCl3).
Mechanism of Production of ROS
- In all cells (Fig. 1.11): ROS are produced normally in small amounts in the mitochondria during the reduction-oxidation (redox) reactions occurring during mitochondrial respiration and production of energy.
- Superoxide (O−•2) is converted to hydrogen peroxide (H2O 2) spontaneously and by the action of the enzyme superoxide dismutase (SOD).
- Hydrogen peroxide (H2O2) in the presence of metals (e.g. Fe2+) is converted by Fenton reaction to a highly reactive free radical called hydroxyl radical (•OH).
- Superoxide (O−•2) is also converted to peroxynitrite (ONOO−) in the presence of nitric oxide (NO).
- In phagocytic leukocytes (Fig. 1.12): ROS produced to destroy the ingested microbes and other substances produced during inflammation.
- During phagocytosis ROS produced in the phagosomes and phagolysosomes is formed in the leukocytes (mainly neutrophils and macrophages) by a process similar to mitochondrial respiration. This process is called as respiratory burst.
- Superoxide (O−•2) is synthesized via NADPH oxidase (nicotinamide adenine dinucleotide phosphate/respiratory burst oxidase) (phagocyte oxidase) present in the phagosome and phagolysosomal membrane of the leukocytes.
- Superoxide (O−•2) is converted to hydrogen peroxide (H2O2).
- Hydrogen peroxide (H2O2) in the presence of myeloperoxidase enzyme is converted to highly reactive compound hypochlorite (HOCl).
Conditions Associated with Increased Generation of Oxygen-derived Free Radicals (Fig. 1.11)
- During inflammation and microbial killing by phagocytes.
- Drugs and chemical injury, including chemical carcinogens.
- Radiation injury (e.g. ultraviolet light, X-rays).
- Reduction-oxidation reactions.
- Ischemia-reperfusion injury induced by restoration of blood flow in ischemic tissue.
- Transition metals such as iron and copper donate or accept free electrons during intracellular reactions and catalyze free radical formation, as in the Fenton reaction (H2O2 + Fe2+ → Fe3+ + OH + OH−).
- Cellular aging.

Mechanisms of Removal/Neutralization of Free Radicals (Fig. 1.11)
Q. Write short note on antioxidants.
Serum, tissue fluids and host cells have antioxidant mechanisms, which protect against potentially harmful oxygen-derived radicals (Table 1.2). These include:
- Spontaneous decay
- Free radical–scavenging systems.
- Enzyme catalase neutralize peroxidase (H2O2) free radicals by converting it into water and oxygen.
- Enzyme superoxide dismutases (SODs) neutralizes superoxide free radicals by converting it into hydrogen peroxide.
- Enzyme glutathione peroxidase (enhances glutathione) neutralizes peroxidase (H2O2), hydroxyl and acetaminophen free radicals.
- Exogenous antioxidants: For example, vitamins E, vitamin A, ascorbic acid and glutathione.
- Endogenous antioxidants: Iron and copper are reactive metals, which can catalyze the formation of ROS. Their activities are minimized by binding of these ions to storage and transport proteins (e.g. transferrin, ferritin and ceruloplasmin).
|
Pathologic Effects of Free Radicals (Fig. 1.11)
15Free radicals can cause cell injury in many diseases. Free radicals can activate both necrosis and apoptosis. Various effects of free radicals are:
- Lipid peroxidation in membranes causes extensive membrane damage.
- Cross-linking and oxidative modification of proteins damages the enzyme activity and causes abnormal folding of proteins.
- Damage to DNA.
Effects of Cell Injury
Defects in Membrane Permeability and Membrane Damage
- Reversible injury: In most forms of cell injury, in the early phase there is selective loss of membrane permeability.
- Irreversible injury: With the obvious membrane damage, the cell cannot return to normal.
Mechanisms of Membrane Damage
- Indirect damage:
- Reactive oxygen species (ROS): It causes injury to cell membranes by lipid peroxidation.
- Decreased phospholipid synthesis: Hypoxia through defective mitochondrial function → decreases the production of ATP by ischemic cells → leads to decreased phospholipid synthesis in all cellular membranes (including the mitochondria) and energy-dependent enzymatic activities.
- Increased phospholipid breakdown: Severe cell injury increases levels of cytosolic and mitochondrial Ca2+ → results in calcium-mediated activation of endogenous phospholipases → which degrades membrane phospholipids → leads to the accumulation of lipid breakdown products → cause membrane damage.
- Cytoskeletal damage: Cytoskeletal filaments connect the plasma membrane to the cell interior. Increased cytosolic calcium activates proteases which may damage the cytoskeletal elements and cell membrane.
- Direct damage: The plasma membrane can also be damaged directly by various bacterial toxins, viral proteins, lytic complement components and a variety of physical and chemical agents.
Consequences of Membrane Damage
Cell injury may damage any membrane, but most important are:
- Mitochondrial membrane damage: It results in:
- Opening of the mitochondrial permeability transition pore leading to decreased ATP.
- Release of proteins that trigger apoptotic death.
- Plasma membrane damage: It leads to loss of:
- Osmotic balance and influx of fluids and ions
- Cellular contents.
- Lysosomal membrane damage: It leads to:
- Leakage of lysosomal enzymes into the cytoplasm
- Activation of lysosomal enzymes → which results in digestion of proteins, RNA, DNA and glycogen → leads to cell death by necrosis.
Damage to DNA and Proteins
- Causes of DNA damage: Exposure to DNA damaging drugs, radiation or oxidative stress.
- Repair mechanism: Cells have mechanisms to repair DNA damage. However, if the damage is too severe to be corrected, the cell initiates a suicide program causing death by apoptosis.
ISCHEMIA-REPERFUSION INJURY
Q. Write short note on ischemia-reperfusion injury.
- Decreased blood flow to a tissue or organ is called ischemia.
- Depending on the severity and duration of ischemia, the involved tissue may adapt, undergo injury (reversible), or die (irreversible). Therapies to restore blood flow is an important modality of treating ischemia.
- If the involved cells of the tissue are reversibly injured, the restoration of blood flow (reperfusion) often beneficial. However, under certain circumstances the restoration of blood flow to cells that have been ischemic (reversibly injured) but have not died (irreversibly injured), can paradoxically exacerbate and produce injury at an accelerated pace.
- The damaging process is set in motion during reperfusion and reperfused tissues undergoes loss of cells (new damage) in addition to the cells that are irreversibly damaged (died) at the end of ischemia. This damaging process is called as ischemia-reperfusion injury.
- Clinical importance: It contributes to tissue damage following reperfusion in myocardial infarction and cerebral infarction.
Mechanism of Reperfusion Injury
New damage may be initiated during reoxygenation includes:
- Increased generation of reactive oxygen and nitrogen species:
- Increased production of free radicals: They may be produced from parenchymal and endothelial cells and from infiltrating leukocytes in reperfused tissue as a result of mitochondrial damage, causing incomplete reduction of oxygen, or because of the action of oxidases in leukocytes, endothelial cells, or parenchymal cells.
- Decreased antioxidant mechanism: Ischemia may result in defective cellular antioxidant defense mechanisms, favoring the accumulation of free radicals.
- Inflammation: Ischemic injury produces cytokines and increased expression of adhesion molecules by hypoxic parenchymal and endothelial cells. They recruit circulating neutrophils to reperfused tissue causing inflammation. The inflammation causes further tissue injury.
- Activation of the complement system: It is an important mechanism of immune-mediated injury. Some IgM antibodies may get deposited in ischemic tissues. When blood flow is restored, complement proteins may bind to the deposited antibodies and complement system may be activated → cause inflammation and more injury to cells.
TYPES OF CELL INJURY
Two types: Reversible and irreversible. Reversible injury may progress to a reversible stage and result in cell death.
Reversible Cell Injury
If the stimulus is acute and brief or mild, the cell injury produces changes in the cells which are reversible up to a certain point.
Light microscope features of reversible cell injury: Two patterns of reversible cell injury namely cellular swelling and fatty change.
- Cellular (hydropic) swelling: It is due to changes in ion concentrations and fluid homeostasis. There is increased flow of water into the cells and results in increased water content of injured cells.
- Steatosis (fatty change) explained above.
Steatosis (Fatty Change)
Q. Write short note on causes, pathogenesis and morphology of fatty/steatosis liver. Add a note on special stains for fat.
Abnormal accumulations of triglycerides within cytosol of the parenchymal cells.
Organs involved: Seen in organs involved in fat metabolism namely liver. It may also occur in heart, muscle and kidney.
Causes
- Disorders with hepatocyte damage: Alcoholic abuse, protein malnutrition, starvation, anoxia (anemia, cardiac failure), toxins (carbon tetrachloride, chloroform, etc.) and Reye syndrome. Alcohol is the most common cause of fatty change in the liver.
- Disorders with hyperlipidemia: Obesity, diabetes mellitus or congenital hyperlipidemia.
Pathogenesis of Fatty Liver
Various mechanisms are involved in excess accumulation of triglyceride in the liver and one or more mechanism may be responsible.
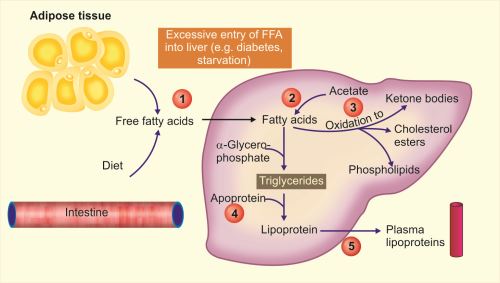
- Excessive entry of free fatty acids (FFA) into the liver(1 in Fig. 1.13): From peripheral stores FFA enters into liver during starvation and diabetes.
- Defective metabolism of lipids: This may be due to:
- Increased synthesis of fatty acids by liver (2 in Fig. 1.13).
- Decreased oxidation of fatty acids into ketone bodies (3 in Fig. 1.13) resulting in increased esterification of fatty acids into triglycerides.
- Decreased synthesis of apoproteins (e.g. in CCl4 and protein malnutrition) causes decreased formation of lipoproteins from triglycerides (4 in Fig. 1.13).
- Defective excretion of lipoproteins: Fatty liver may also develop due to defect in excretion of lipoproteins from liver into the blood (5 in Fig. 1.13).
Heart
Q. Write short note on heart in fatty change.
Lipid in the cardiac muscle can have two patterns:
- Alternate involvement: Prolonged moderate hypoxia (e.g. severe anemia), create grossly apparent bands of involved yellow myocardium alternating with bands of darker, red-brown, uninvolved myocardium (tigered effect, tabby cat appearance).
- Uniform involvement: More severe hypoxia or some types of myocarditis (e.g. diphtheria infection) show more uniform involvement of myocardial fibers.
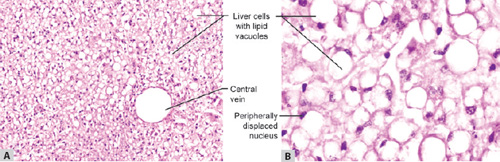
Figs 1.15A and B: (A) Fatty liver in which the hepatocytes show accumulation of fat which appear as clear vacuole in the cytoplasm; (B) Hepatocytes at higher magnification in which the nucleus is displaced to the periphery by accumulated fat
Cholesterol Deposits
Intracellular accumulation of cholesterol or cholesterol esters in macrophages may occur when there is hypercholesterolemia. It appears microscopically as intracellular.
Atherosclerosis
It is a disease of aorta and large arteries characterized by the presence of atherosclerotic plaques composed of smooth muscle cells and macrophages within the intima filled with lipid vacuoles. Most of the lipid is cholesterol and cholesterol esters (refer Chapter 14).
Xanthoma
Q. Write short note on xanthoma.
Intracellular accumulation of cholesterol within macrophages is found in acquired and hereditary hyperlipidemic 18 states. The tumor mass produced by the macrophages filled with cholesterol is termed xanthomas. Microscopically, it consists of clusters of foamy cells in the subepithelial connective tissue of the skin and in tendons.
Irreversible Cell Injury
If the cell is exposed to continuous injurious stimulus or if the injury is severe, the cells undergo cell death. Two main types of cell death: Necrosis and apoptosis.
- Necrosis: Always a pathologic process (refer below).
- Apoptosis: May be physiological or pathological (refer page 22).
NECROSIS
Q. Define necrosis. Describe the various types of necrosis, causes and pathology of each with suitable examples.
Definition: Morphological changes indicative of cell death in a living tissue following harmful injury. Necrosis is an “accidental” and unregulated form of cell death. It results from damage to cell membranes and loss of ion homeostasis. The necrotic cells cannot maintain integrity of membrane and their contents leak out. This bring out acute inflammatory reaction in the surrounding tissue.
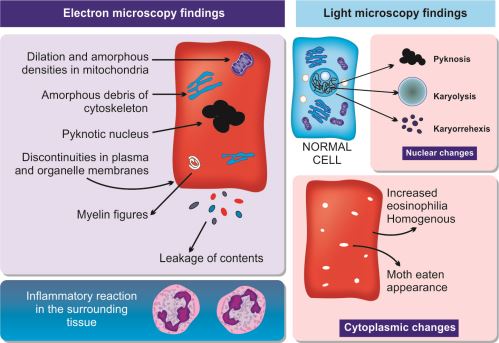
Electron microscopic findings of necrosis are diagrammatically shown in Figure 1.16.
Patterns/Types of Tissue Necrosis
Coagulative Necrosis
Q. Write short note on coagulative necrosis.
Common type, outline of dead tissues is preserved (at least for few days). Infarct is a localized area of coagulative necrosis.
- Causes: Ischemia caused by obstruction in a vessel.
- Mechanism: Ischemia denatures and coagulates structural proteins and enzymes.

Figs 1.17A to C: (A) Gross appearance of infarct of kidney; (B) Microscopy of normal kidney parenchyma; (C) Infarcted area of kidney

Figs 1.18A and B: Microscopic appearance of an abscess consisting of liquified necrotic cell debris and dead/disintegrating neutrophils. (A) Hematoxylin and eosin; (B) Diagrammatic appearance of brain abscess
Liquefactive Necrosis (Colliquative Necrosis)
Q. Write short note on liquefactive/colliquative necrosis.
Dead tissue rapidly undergoes softening and transforms into a liquid viscous mass.
- Causes:
- Ischemic injury to central nervous system (CNS)
- Suppurative infections: Infections by bacteria which stimulate the accumulation of leukocytes.
- Mechanism: Liquefaction is due to digestive action of the hydrolytic enzymes released from dead cells (autolysis) and leukocytes (heterolysis).

Caseous Necrosis
Q. Write short note on caseous necrosis.
Distinctive type of necrosis which shows combined features of both coagulative and liquefactive necrosis.
- Cause: Characteristic of tuberculosis and is due to the hypersensitivity reaction.

Fat Necrosis
Q. Write short note on fat necrosis.
It refers to focal areas of fat destruction, which affects adipose tissue.
Types:
- Enzymatic fat necrosis: Occurs in adipose tissue around acutely inflamed pancreas (in acute pancreatitis).
- Mechanism: In pancreatitis, the enzymes (one of them is lipase) leak from acinar cells and causes tissue damage. Lipase destroys fat cells and liberates free fatty acids which combine with calcium and form calcium soaps (fat saponification).
- Traumatic fat necrosis: Occurs in tissues with high fat content (like in breast and thigh) following severe trauma.
Fibrinoid Necrosis
Characterized by deposition of pink-staining (fibrin-like) proteinaceous material in the tissue matrix with a staining pattern reminiscent of fibrin (Figs 1.22 and 14.10). It obscures the underlying cellular detail.
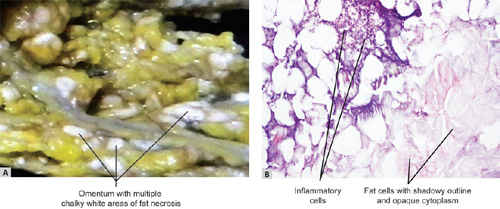
Figs 1.21A and B: (A) Omentum shows multiple chalky white areas of fat necrosis caused by acute pancreatitis; (B) Fat necrosis shows necrotic fat cells in the right lower part and inflammatory reaction between normal (left upper area) and area of fat necrosis
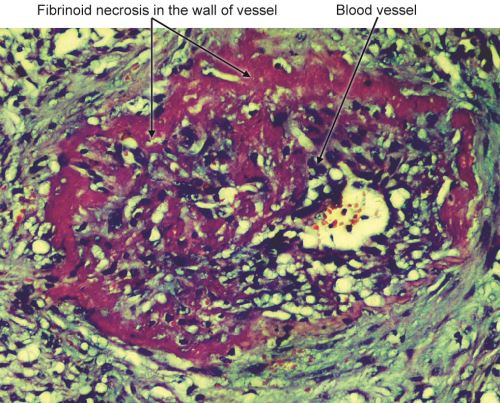
- Causes: Usually seen in immune-mediated (deposition of antigen-antibody complexes in the wall of vessels) vascular injury/vasculitis (e.g. polyarteritis nodosa), malignant hypertension, Aschoff bodies in rheumatic heart disease, placenta in preeclampsia, or hyperacute transplant rejection.
Gangrene (Gangrenous Necrosis)
Q. Define gangrene. Mention its types and differences between them.
It is massive necrosis with superadded putrefaction.
Types: Two types, namely dry and wet gangrene. A variant of wet gangrene known as gas gangrene is caused by clostridia (Gram-positive anaerobic bacteria).
Dry Gangrene
- Causes: Arterial occlusion (e.g. atherosclerosis).
- Sites: It usually involves a limb, generally the distal part of lower limb (leg, foot, and toe).

Fig. 1.23:
Dry gangrene of left leg shows dry shrunken discolored gangrenous foot separated from adjacent normal area by a line of demarcation
Differences Between Dry and Wet Gangrene (Table 1.3)
Q. List the differences between dry and wet gangrene.
Gas gangrene: Special type of wet gangrene caused by infection with a gas forming anaerobic clostridia. These organisms enter into the tissues through open contaminated wounds (e.g. muscles, complication of operative procedures on colon). Toxins produced by them cause local necrosis and edema and are also absorbed causing severe systemic manifestations.
Gummatous Necrosis
The necrotic tissue is firm and rubbery and is usually found in syphilis.
| |||||||||||||||||||||||||||||||||||||
Ultrastructural differences between reversible and irreversible injury is presented in Table 1.4.
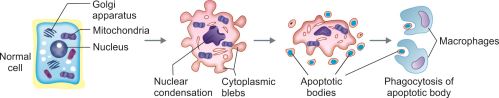
APOPTOSIS
Q. Write short note on apoptosis.
Apoptosis is a type of (programmed) cell death induced by a tightly regulated suicide program. It is characterized by activation of intrinsic enzymes of the cell that degrade its own nuclear DNA and proteins (nuclear and cytoplasmic).
Causes of Apoptosis
Physiological Situations
- Removal of excess cells during embryogenesis and developmental processes: For example, disappearance of web tissues between fingers and toes.
- Elimination of cells after withdrawal of hormonal stimuli: For example, endometrial cell breakdown during the menstrual cycle.
- Elimination of cells after withdrawal of tropic stimuli: For example, neutrophils in an acute inflammatory response, lymphocytes after immune response.
- Elimination of potentially harmful cells: In immunology, the clones of self-reactive lymphocytes that recognize normal self antigens are deleted by apoptosis.
Pathological Conditions
Apoptosis eliminates cells that are genetically altered or damaged beyond repair. It is responsible for cell loss in many pathologic states:
- Elimination of cells with damaged DNA: DNA may be damaged by many injurious agents like radiation, cytotoxic anticancer drugs and hypoxia.
- Mainly tumor-suppressor gene p53 recognizes cells with damaged DNA and assesses whether it can be repaired. If the damage is too severe to be repaired, p53 triggers apoptosis.
- Destroying cells with dangerous mutations or with DNA damage beyond repair by apoptosis prevents the development of cancer.
- In certain cancers, where p53 is mutated or absent, the apoptosis is not induced in cells with damaged DNA.
- Elimination of cells with excessively accumulated misfolded proteins: Mutations in the genes encoding proteins or extrinsic factors (damage due to free radicals) may result in accumulation of unfolded or misfolded proteins.
- Excessive intracellular accumulation of these abnormally folded proteins in the ER is known as ER stress, which results in apoptotic cell death.
- Apoptosis caused by the accumulation of misfolded proteins is found in several degenerative diseases of the central nervous system (Alzheimer, Huntington, and Parkinson diseases) and other organs.
- Killing of viral infected cells: In viral infections, the infected cells are lost mainly due to apoptosis induced either by the virus (as in adenovirus and HIV infections) or by host human response by cytotoxic T lymphocytes (as in viral hepatitis).
- Elimination of neoplastic cells/rejection of transplant: The T-cell-mediated mechanism is responsible for apoptosis in tumors and cellular rejection of transplants.
- Elimination of parenchymal cells in pathologic atrophy: Obstruction of duct in the parenchymal organs like pancreas, parotid gland and kidney can lead to apoptosis of the parenchymal cells.
Mechanisms of Apoptosis
Q. Write short note on mechanism of apoptosis.
The survival or apoptosis of many cells depends upon balance between two opposite sets of signals namely (1) death signal (proapoptotic) and (2) prosurvival (anti-apoptotic) signals. Unlike necrosis, apoptosis engages the cell's own signaling cascades and results in its own death (suicide). Apoptosis results from activation of enzymes called as caspases (i.e. they are cysteine proteases that cleave proteins after aspartic residues).
Phases of Apoptosis
Divided into (A) initiation phase and (B) execution phase.
A. Initiation phase
Apoptosis is initiated by signals derived from two distinct pathways activated by distinct stimuli, namely (1) intrinsic or mitochondrial pathway and (2) extrinsic or death receptor pathway.
- Intrinsic (mitochondrial) pathway of apoptosis (Fig. 1.25): It is activated by intracellular signals.
- Role of mitochondria in apoptosis:
- Mitochondrial damage is the major mechanism in a variety of physiological and pathological apoptosis.
- Mitochondria contain proteins capable of inducing apoptosis. These include: cytochrome c and several proapoptotic proteins.
- Survival or apoptosis of cell is determined by permeability of mitochondria.
- Mitochondrial permeability is controlled by Bcl 2 family of more than 20 proteins. This family is named after Bcl2, which was identified as an oncogene in a B-cell lymphoma. These proteins may be broadly divided into proapoptotic or anti-apoptotic (prosurvival).
- Proapoptotic proteins: BAX and BAK
- Antiapoptotic proteins: BCL2, BCL-XL, and MCL1. They prevent leakage of mitochondrial proteins that trigger apoptosis. Growth factors and other survival signals stimulate production of antiapoptotic proteins.If the balance shifts to proapoptotic proteins, the apoptotic cascade is activated.
- Causes of mitochondrial injury: The proapoptotic signals include:
- Deprivation/withdrawal of growth factor or survival signals.
- DNA damage by radiation, cytotoxic anticancer drugs, hypoxia either directly or through free radical.
- Accumulation of excessive amount of misfolded proteins (endoplasmic reticulum stress).
- Increased intracellular free calcium.
- Steps in intrinsic (mitochondrial) pathway: Mitochondrial injury causes increased mitochondrial permeability and release proapoptotic molecules (death inducers) into the cytoplasm. The different steps are as follows:
- The above mentioned causes of mitochondrial injury activate a number of sensors of BCL2 family called BH3-proteins. They in turn activate two critical proapoptotic Bcl2 family effector proteins, namely Bax and Bak.
- Bax and Bak create channels in the mitochondria that allow release of several mitochondrial proteins from the inner mitochondrial membrane to leak out into the cytosol (cytoplasm).
- One of these proteins is cytochrome c which binds to a protein called apoptosis-activating factor-1 (Apaf-1) and forms an important caspase cascade activator called apoptosome. 25This complex binds to caspase-9, the critical initiator caspase of the mitochondrial pathway which sets in an auto-amplification process.
- The cytoplasm of the normal cells contains proteins which block the activation of caspases and function as physiologic inhibitors of apoptosis (called IAPs). Other mitochondrial proteins may enter the cytoplasm and neutralize these IAPs and initiate caspase cascade.
- Sensors of Bcl2 family namely BH3-only proteins also bind to and block the function of protective antiapoptotic proteins namely Bcl2and Bcl-xL.
- 2. Extrinsic (death receptor–initiated) pathway of apoptosis (Fig. 1.25)
- This pathway is initiated by extracellular signals.
- Many cells express “death-receptors” molecules on the surface of plasma membrane that trigger apoptosis. Death receptors are member of the TNF (tumor necrosis factor) receptor family that contain a cytoplasmic domain called the death domain because it is essential for delivering apoptotic signals.
- In the extrinsic (death receptor) pathway, apoptosis is initiated when the death receptors present gets activated.
- The well-known death receptors are the type 1 TNF receptor (TNFR1) and a related protein called Fas (CD95). Fas death receptor is expressed on many cell types and the binding ligand for Fas is called Fas ligand (FasL/CD95L).
- Functions of extrinsic pathway: This pathway is involved in eliminating:
- Self-reactive lymphocytes thereby avoiding autoimmunity. FasL is expressed on T-cells that recognize self-antigens and function to eliminate self-reactive lymphocytes.
- Virus infected cells through cytotoxic T lymphocytes.
- Tumor cells through cytotoxic T lymphocytes.
- Steps in extrinsic pathway:
- Extrinsic pathway become activated when CD95/Fas binds to its ligand CD95L/FasL.
- When FasL binds to Fas receptors, their cytoplasmic death domains binds with an adapter protein. This adapter protein also contains a death domain and is called Fas-associated death domain (FADD).
- FADD in turn binds to pro-caspase-8 (an inactive form of caspase-8) via a death domain and generate active caspase-8.
- Activated caspase-8 mediate the execution phase of apoptosis.
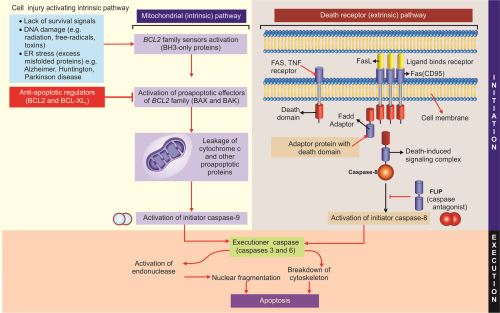
B. Execution Phase of Apoptosis (Fig. 1.25)
- The above mentioned two initiating pathways produce initiator caspases namely: (1) the mitochondrial pathway activates initiator caspase-9, and (2) the death receptor pathway activates the initiator caspase-8.
- The initiator caspases activate another series of caspases called executioner caspases (such as caspase-3 and −6) that mediates the final phase of apoptosis.
- Executioner caspases act on many cellular components and activate DNase, which induces fragmentation of nuclei.
- Caspases also degrade components of nuclear matrix and cytoskeleton resulting in fragmentation of involved cells.
Removal of Apoptotic Cells
Apoptosis is a regulated mechanism of cell death with the least possible reaction by host.
- Phagocytosis: Apoptotic cells and bodies are engulfed and removed by phagocytic cells (mainly macrophages). The phagocytosis is so efficient that these dead cells and apoptotic bodies disappear within minutes. Even when the apoptosis is extensive their rapid removal prevents release of their cellular contents which may elicit inflammation.
- Factors favoring phagocytosis: The apoptotic cells and apoptotic bodies undergo several changes in their membranes and produce signals that favor phagocytosis of these cells/bodies.
- Expression of phosphatidylserine: In healthy cells, phosphatidylserine is present on the inner leaflet of the plasma membrane. In cells undergoing apoptosis phosphatidylserine turns out and is expressed on the outer layer of the membrane causing easy recognition by receptors present on the macrophage.
- Secretion of soluble factors: Apoptotic cells secrete soluble factors (e.g. thrombospondin) that recruit phagocytes.
- Natural antibodies and proteins of the complement system may coat apoptotic bodies which aids in phagocytosis.
Diagnosis/Detection of Apoptosis
- DNA fragmentation assay is carried out by electrophoresis of genomic DNA. Apoptosis produces “step ladder pattern” in contrast to smeared pattern seen in necrosis.
- Terminal deoxynucleotidyl transferase biotin d-UTP Nick End Labeling (TUNEL) technique for in vivo detection of apoptosis.
- Chromatin condensation seen by hematoxylin and eosin, Feulgen and acridine orange staining.
- Estimation of:
- Cytosolic cytochrome c
- Activated caspase
- Annexin V: Apoptotic cells express phosphatidylserine on the outer layer of plasma membrane because of which these cells are recognized by the dye Annexin
- Propidium iodide assay by flow cytometry/fluorescent microscopy.
Disorders Associated with Dysregulated Apoptosis
- Disorders with reduced apoptosis: It may allow the survival of abnormal cells.
- Cancer
- Autoimmune disease.
- Disorders with increased apoptosis: This will cause an excessive loss of cells.
- Neurodegenerative diseases (Alzheimer, Huntigton, Parkinson disease).
- Ischemic injury: In myocardial infarction and stroke.
- Death of virus-infected cells: Many viral infections, important being acquired immune deficiency syndrome (AIDS).
Clinical Significance of Apoptosis in Cancers
- Normally, cells with damaged (mutated) DNA are cleared in the body by undergoing apoptosis.
- Apoptosis may be reduced in some cancers. Best established role of Bcl2 in protecting tumor cells from undergoing apoptosis is observed in follicular lymphoma. In this type of non-Hodgkin lymphoma of B cell origin, there is translocation (14; 18) (q32; q21) which causes over expression of antiapoptotic protein Bcl2. This in turn increases the BCL2/Bcl-xL buffer, protecting abnormal B lymphocytes from undergoing apoptosis and allows them to survive for long periods.
Q. List the differences between apoptosis and necrosis.
Differences between apoptosis and necrosis are summarized in Table 1.5.
- Necroptosis: It is a type of cell death that shows features of both necrosis and apoptosis. It is caspase-independent. It resembles morphologically necrosis and mechanistically apoptosis.
- Pyroptosis: It is a type of programmed cell death accompanied by the release of fever producing cytokine IL-1 and bears some biochemical similarities with apoptosis.
- Autolysis (means self-lysis) is destruction of the cell by its own hydrolytic enzymes released from lysosomes. Autolysis is generally reserved for postmortem change. It develops rapidly in some tissues rich in hydrolytic enzymes such as pancreas and gastric mucosa. It occurs little slowly in tissues such a the heart, liver and kidney; and slow in fibrous tissue. Microscopically, the cellular details are loss and they appears as cells with homogeneous and eosinophilic cytoplasm.
Overview of cell injury in presented in Figure 1.26.
PATHOLOGIC CALCIFICATION
Q. Write short note on pathologic calcification.
Abnormal deposition of calcium salts in tissues other than osteoid or enamel. It is also associated with deposition of small amounts of iron, magnesium and other minerals.
Types of pathologic calcification are: (1) dystrophic and (2) metastatic.
Dystrophic Calcification
Q. Write short note on dystrophic calcification.
Deposition of calcium salts in dying or dead tissues.
Causes
- Necrotic tissue: Calcification in caseous, enzymatic fat necrosis, in dead eggs of Schistosoma, cysticercosis and hydatid cysts.
- Degenerating tissue:
- Heart valves: Occurs in aging or damaged heart valves
- Atherosclerosis, goiter of thyroid, dense old scar, cysts (e.g. epidermal and pilar cysts of skin).
- Monckeberg's medial calcific sclerosis: Calcification in the media of the muscular arteries (Fig. 1.27A) in old people.
- Psammoma bodies: Single necrotic cells on which several layers of mineral get deposited progressively to create lamellated shape called psammoma bodies (Fig. 1.27B).
Metastatic Calcification
Q. Write short note on metastatic calcification.
Deposition of calcium salts in apparently normal tissues. It is associated with hypercalcemia secondary to deranged calcium metabolism.
Causes
- Increased secretion of parathyroid hormone (PTH) with subsequent bone resorption-hyperparathyroidism.
- Destruction of bone tissue: Secondary to primary tumors of bone marrow (e.g. multiple myeloma, leukemia and metastatic tumors to bone).
- Vitamin D–related disorders: Vitamin D intoxication.
- Renal failure: Causes retention of phosphate, leading to secondary hyperparathyroidism.
- Others: Sarcoidosis and milk alkali syndrome.

Sites
- Lungs: Alveolar septa of the lung.
- Kidney: Basement membrane of the renal tubules.
- Blood vessels: On the internal elastic lamina of systemic arteries and pulmonary veins.
- Stomach: Interstitial tissues of the gastric mucosa.
Gross: Appear as fine, white granules or clumps, feels gritty and sand-like.
Microscopy: Basophilic, amorphous granular (Fig. 1.27), clumped appearance.
HYALINE CHANGE
Q. Write short note on hyaline change.
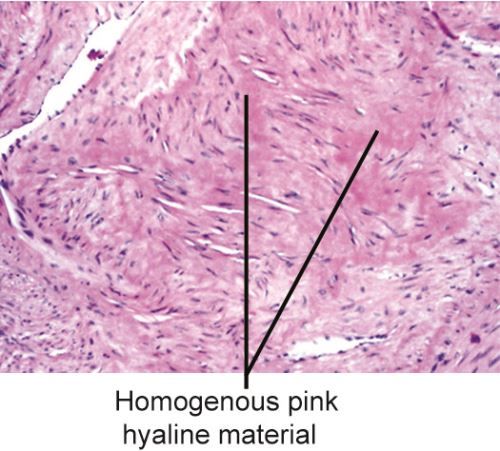
Hyaline refers to an alteration within cells or in the extracellular space, which gives a homogeneous, glassy, pink appearance in routine histological sections.
Causes (Table 1.6)
Intracellular Hyaline
-
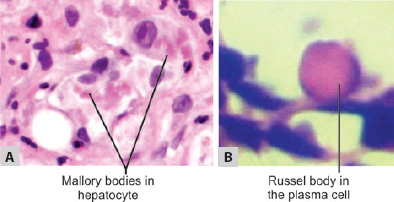
TABLE 1.6 Examples of hyaline change Intracellular hyalineExtracellular hyaline1. Mallory bodies2. Russell bodies (e.g. multiple myeloma)3. Crooke's hyaline4. Zenker's hyaline change1. Collagenous fibrous tissue in scar2. Hyaline change in uterine leiomyoma3. Hyaline membrane in newborn4. Hyaline arteriosclerosis5. Hyalinization of glomeruli in chronic glomerulonephritis6. Corpora amylacea in prostate, brain, spinal cord in elderly, old infarct of lung - Russell bodies are excessive accumulation of immunoglobulins in the rough endoplasmic reticulum of plasma cells (Fig. 1.28B).
- Zenker's degeneration: Hyaline degeneration of rectus abdominalis muscle (becomes glassy and hyaline) in typhoid fever.
Extracellular Hyaline
Q. Write short note on Russel bodies.
- Collagenous fibrous tissue in old scars.
- Hyaline change in uterine leiomyoma (Fig. 1.29).
- In chronic glomerulonephritis, the glomeruli show hyalinization.
PIGMENTS
Q. Write short note on various pigments.
Pigments are colored substances, which are either normal constituents of cells (e.g. melanin), or are abnormal and accumulate in cells. Different types of pigments are listed in Table 1.7.
|
Melanin
Melanin is an endogenous, brown-black, non-hemoglobin-derived pigment. It is produced by the melanocytes and dendritic cells by the oxidation of tyrosine to dihydroxyphenylalanine by the enzyme tyrosinase. It is stored as cytoplasmic granules in the phagocytic cells namely melanophores. Normally, it is present in the hair, skin, mucosa at some places, choroid of the eye, meninges and adrenal medulla. Various disorders of melanin pigmentation produce generalized and localized hyperpigmentation and hypopigmentation (Table 1.8).
|
Alkaptonuria
Homogentisic acid is a pathological black pigment formed in rare metabolic autosomal recessive disorder termed alkaptonuria. It is characterized by deficiency of an oxidase enzyme needed for breakdown of homogentisic acid. This leads to accumulation of homogentisic acid pigment in the skin, connective tissue, cartilage, capsules of joints, ligaments and tendons. The pigment is melanin-like and the pigmentation is known as ochronosis. The homogentisic acid is excreted in the urine (homogentisic aciduria). The urine of patients of alkaptonuria, if allowed to stand for some hours in air, turns black due to oxidation of homogentisic acid.
Hemosiderin
Q. Write short note on hemosiderin and hemosiderosis.
It is a hemoglobin-derived, golden yellow-to-brown, granular or crystalline pigment and is one of the major Russel bodies storage forms of iron.
Causes
Local or systemic excess of iron cause hemosiderin to accumulate within cells.
- Local excesses:
- Bruise
- Brown induration of lung in chronic venous congestion of lung (refer Fig. 5.1).
- Systemic excesses: Systemic overload of iron is known ashemosiderosis. The main causes:
- Increased absorption of dietary iron.
- Excessive destruction of red cells: For example, hemolytic anemias.
- Repeated blood transfusions.
Other Pigments
- Hemochromatosis: Severe accumulation of iron is associated with damage to liver, heart and pancreas. The triad of cirrhosis of liver, diabetes mellitus (due to pancreatic damage) and brown pigmentation of skin constitute bronze diabetes.
- Hemozoin: It is a brown-black pigment containing heme in ferric form. This pigment is seen in chronic malaria and in mismatched blood transfusions.
- Bilirubin is the normal major pigment found in bile. It is non-iron containing pigment derived from hemoglobin.
- Lipofuscin
Q. Write short note on lipofuscin and brown atrophy of heart.
- Lipofuscin is an insoluble golden-brown endogenous pigment. It also called as lipochrome or wear and tear pigment.
- Composition: It is composed of mixture of lipids, phospholipids and proteins. It is accumulated by accretion of peroxidized unsaturated lipids and oxidized cross-linked proteins. The term lipofuscin is derived from the Latin (fuscus, brown), and refers to brown lipid.
- Significance: It indicates a product of free radical injury and lipid peroxidation. Lipofuscin does not injure cell or its functions. It is observed in cells undergoing slow, regressive changes and is particularly prominent in the liver and heart (often called brown atrophy of heart) of aging patients or patients with severe malnutrition and cancer cachexia.
- Appearance: Microscopically, it appears as a yellow-brown, finely granular cytoplasmic pigment, often present in the perinuclear region.
Commonly used histochemistry (special stains) in histopathology are listed in Table 1.9.
CELLULAR AGING
Definition of aging: It is the gradual, insidious and progressive declines in structure and function (involving molecules, cells, tissues, organs and organisms) that begin to unfold after the achievement of sexual maturity.

Cellular aging begins from conception and continues till death. With aging physiological and structural changes develop in almost all systems. There is progressive loss of functional capacity.
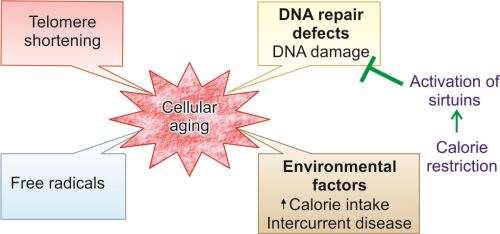
Causes
Aging is multifactorial and is affected by genetic factors and environmental factors.
- Genetic abnormalities: It causes progressive decline in cellular function and viability.
- Environmental factors: These include diet, social conditions and development of age-related diseases (e.g. atherosclerosis, diabetes and osteoarthritis). They cause progressive accumulation of sublethal injury over the years at cellular and molecular level.
- Cellular aging may lead to death of the cell or decreased capacity of cells to respond to injury and increasing difficulties in maintaining physiological homeostasis.
Mechanism of Cellular Aging
Decreased Cellular Replication
Most normal cells have a limited capacity for replication. After about 60–70 cell divisions, all cells become arrested in a terminally nondividing state, known as senescence. Werner syndrome is a rare disease characterized by premature aging, damaged DNA and a markedly reduced capacity of cells to divide (shortening of telomere). The following mechanisms may be responsible for progressive senescence of cells and decreased cellular replication in aging.
Telomere Shortening
Telomeres are protective, short repeated sequences of DNA (TTAGGG) present at the end regions of chromosomes. Telomeres ensure the complete copying of chromosomal ends during the S-phase of the cell cycle. With each cell division in somatic cells, a small section of the telomere is not duplicated and telomeres become progressively shortened (Fig. 1.30). When telomeres are sufficiently shortened, cells stop dividing leading to a terminally nondividing state. Telomeres represent a ‘biological clock’, which prevents uncontrolled cell division and cancer. Telomere shortening may be one of the mechanisms responsible for decreased cellular replication.
Telomerase
Telomerase is an enzyme that regenerates and maintains telomere length. Telomerase is absent in most of the somatic cells. Germ cells have high telomerase activity and thus they have extended replicative capacity (Fig. 1.30). In cancers, the telomerase may be reactivated in tumor cells resulting in maintenance of length of telomeres. It may be an essential step in formation of cancer.
Accumulation of Metabolic and Genetic Damage (Fig. 1.31)
Lifespan of the cell is determined by a balance between cumulative metabolic damage and counteracting repair responses.
Metabolic Damage
Reactive oxygen species: One of the toxic products that cause damage to the cells is free radical mainlyreactive oxygen species (ROS). ROS may be either produced in excess, or there is reduction of antioxidant defense mechanisms (refer page 13–15).
- Excessive production of ROS may be due to environmental influences (ionizing radiation) and mitochondrial dysfunction.34
- Reduction of antioxidant defense mechanisms may occur with age (e.g. vitamin E, glutathione peroxidase).
The oxidative damage may be an important cause of senescence in aging. Free radicals may damage DNA, causing breaks and genome instability. Damaged cellular organelles also accumulate as the cells age.
Defective Repair Mechanism
Many protective repair responses counterbalance the metabolic damage in cells. One of them is endogenous DNA repair enzymes, which identify the DNA damage and repairs it. DNA repair mechanisms are defective in diseases such as Werner syndrome and ataxia-telangiectasia.
Thus, aging can be delayed by either by reducing the metabolic damage or by increasing the repair response to that damage.
Factors that Increases Longevity
Caloric Restriction
Calorie restriction prolongs lifespan and this longevity appears to be mediated by a family of proteins known as sirtuin. They have histone deacetylase activity. Red wine can activate sirtuins and thus increase lifespan.
Actions of Sirtuins
- Sirtuins promotes the expression of many genes which increase longevity. The proteins products of these genes increase metabolic activity, reduce apoptosis, stimulate protein folding and inhibit the damaging effects of oxygen-free radicals.
- Sirtuins also increases insulin sensitivity and glucose metabolism.
Growth Factor Signaling
Growth factors, such as insulin-like growth factor trigger the insulin receptor pathway. This results in activation of transcription factors which activate genes that reduce longevity. Mutations in insulin receptor are associated with increased lifespan.